Overview of RNA-seq data
Animal coats play an important role in adaptation to the external environment. Non-hibernating land mammals can adapt to their surrounding conditions through seasonal changes to their coats [7]. Mammalian hair is produced by the proliferation and differentiation of hair follicle cells [8]. The hair follicle, which is the only life-sustaining organ unique to mammals [9], plays an important role in mammalian adaptation to environmental changes. The yak is a rare large livestock animal that lives in alpine and hypoxic environments, and it has cold resistance but no heat resistance; this causes yak to respond very sensitively to temperature differences in different seasons. We selected yak as a model species for our study of hair development, because its hair follicles are in a dynamic relationship with environmental changes, resulting in marked periodic changes in the structure of the hair follicles, and the developmental regulation of this process is very rapid [6].
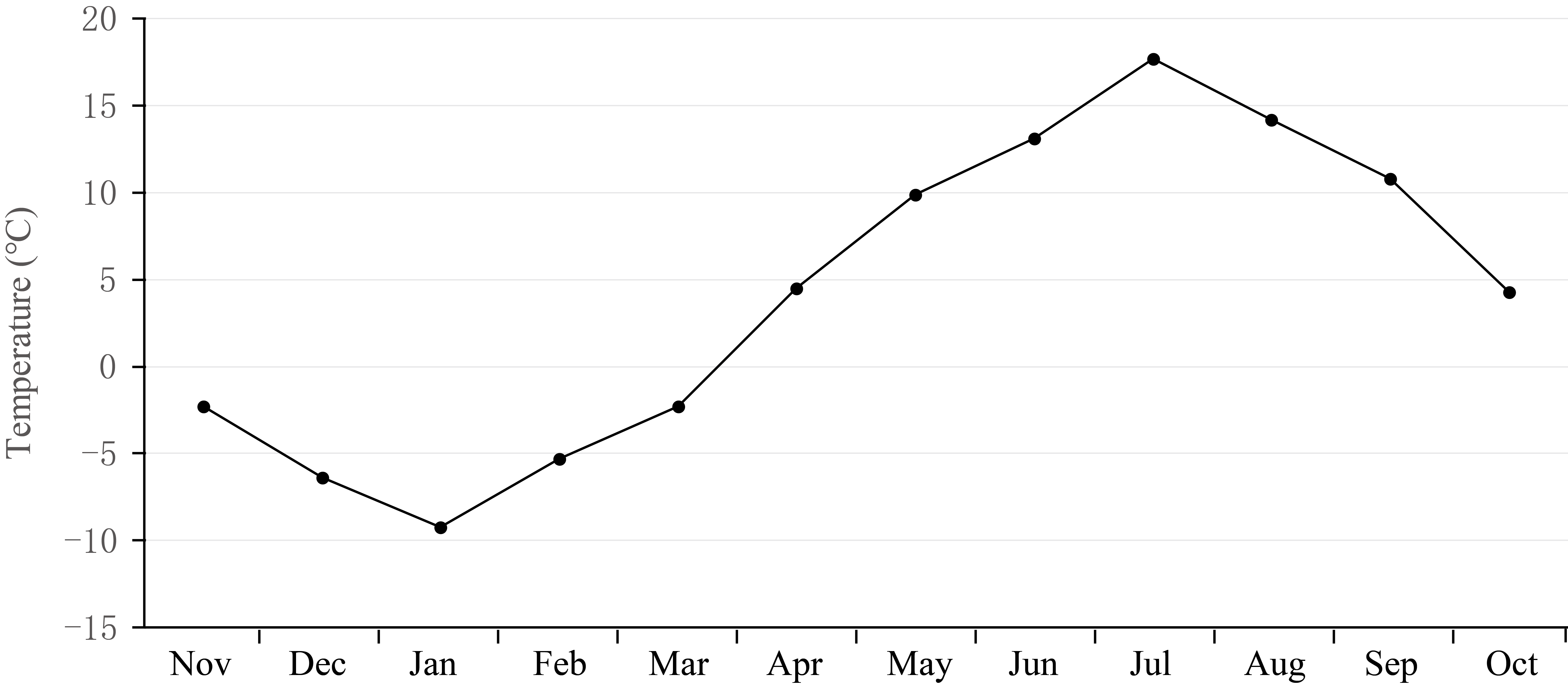
A total of 15 libraries were sequenced from skin follicle tissues of yaks at five different physiological stages (n=3 for each). We selected these particular developmental stages because they are at the key turning points in the year when the temperature changes dramatically, and such temperature changes are one of the important factors affecting the cyclical changes in yak coats (Additional file1: Figure S1). The average amount of raw data for these 15 libraries was 25.15 million reads with 3.94 Gb of raw bases. After discarding low-quality reads, an average of 24.96 million clean reads was obtained, an amount of data sufficient to meet the requirements of this research. The clean reads were mapped to the yak reference genome using Hisat2 software, and the percentage of the total reads mapped was approximately 92.21% for each sample, indicated that the yak reference genome we used was appropriate. Specifically, 91.94%, 92.63%, 92.59%, 91.86% and 92.04% of the clean reads from RNA sampled in Aug, Oct, Jan, Mar and Jun respectively were mapped to the yak reference genome (Additional file2: Table S1).
We used StringTie software to produce more complete and accurate reconstructions of genes and better estimates of expression levels. A gene was considered to be expressed in a given sample if the lower boundary of its FPKM 95% confidence interval was greater than zero [10]. Based on this criterion, 11,666 genes were identified as being expressed in at least one of the 15 samples, and the distribution of FPKM values for these genes was then statistically analyzed. Oesults showed that 90% of the genes were expressed at a medium level (1 ≤ FPKM < 100), less than 8% exhibited high-level expression (FPKM ≥100) and the rest showed low-level expression (FPKM < 1) among the 15 samples analyzed. We found that the proportions of genes at the three expression levels (high, medium, and low) were similar in all samples, indicating that our transcriptome data were consistent and lacked any obvious bias (Figure 1).
Hierarchical clustering and principal component analysis of samples
To assess the consistency of sample collection and explore the transcriptomic relationships among the five stages, we performed hierarchical clustering and principal component analysis (PCA) for these samples using the whole gene expression dataset.
Using hierarchical clustering, we found that the samples were divided into three clusters and that samples from the same period were grouped together, with the Jan samples forming a single cluster, the Aug and Oct samples grouped together and the Mar and Jun samples being another cluster (Figure 2a). In the PCA analysis, the samples collected in the same period were grouped into the same cluster; this is consistent with the results of hierarchical clustering (Figure 2b). Both results showed that the 15 samples could be grouped into three significant phases, and we speculated that these three stages might represent different developmental components of the yak hair cycle. The hair cycle is usually divided into three phases: anagen (growth phase), catagen (regression phase) and telogen (resting phase). Based on the period when yak hair growth is known to occur and on records of mean monthly temperatures, we hypothesized that the Aug and Oct samples were in the anagen of the hair cycle, the Jan samples were in the catagen and the Mar and Jun samples were in the telogen. In the hierarchical clustering plot, we observed that the clustering of the anagen samples was closer to that of the catagen samples than to that of the telogen samples. This result indicated that the development status of the catagen, the dynamic transition between anagen and telogen, was closer to that of the anagen. In addition, we found that one sample from Jun was clustered together with the Mar samples. This phenomenon indicates, on the one hand, that the individual differences between the sampled yaks were relatively large, and on the other hand, that the gene expression patterns of the samples from March and June are very similar.
Change in transcriptome profiles during the hair cycle
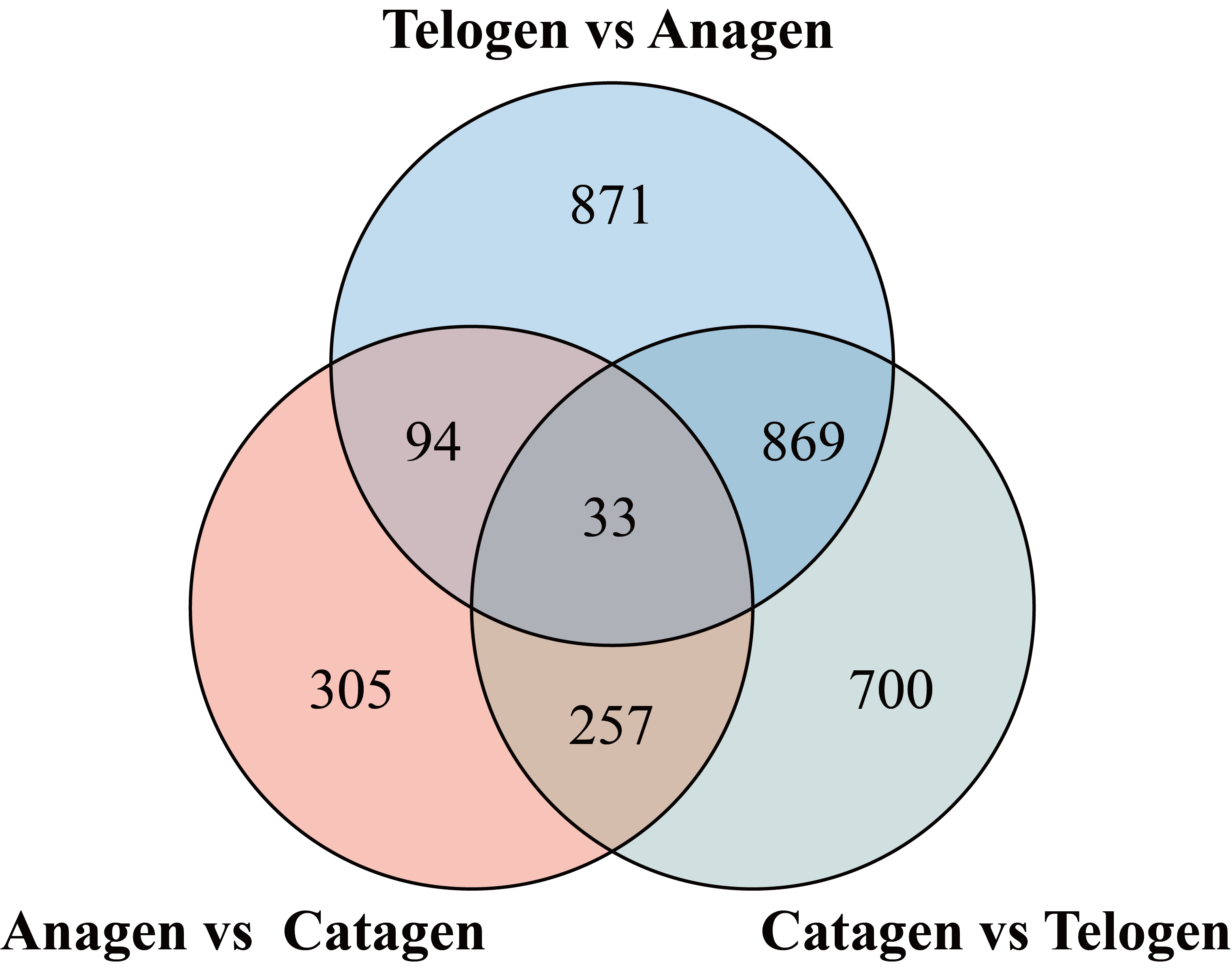
In this study, we used DESeq2 software to identify the genes that were differentially expressed (DEGs) between consecutive stages to determine changes during the hair cycle, resulting in the identification of 2,316 DEGs. Of these DEGs, 714, 534 and 916 genes were upregulated and 1153, 155 and 943 genes were downregulated in telogen versus anagen, anagen versus catagen, catagen versus telogen respectively (Figure 3a). It was clear that, compared to anagen versus catagen, a larger number of genes was up- or down-regulated in telogen versus anagen, and catagen versus telogen, indicating a major change in the transcriptional profile (Additional file 3: Figure S2).
The expression patterns make it clear that there are multitudinous and complex interactions among genes, and genes with similar expression patterns may have similar functions in the hair cycle. An in-depth study of gene expression patterns during the hair cycle may therefore help in gaining a better understanding of the mechanism regulating this cycle. We further investigated the expression patterns of DEGs at three developmental stages during the hair cycle. We classified these 2,316 DEGs into nine gene clusters (C1-C9) based on their expression patterns (Figure 3b), Each cluster represents a unique set of expression patterns.(1) Cluster one: each gene is upregulated in the anagen and downregulated in the other two periods; (2) Cluster two: continuous upregulation from anagen to catagen, but downregulated in telogen; (3) Cluster three: upregulated from anagen to catagen and downregulated in telogen, but mainly expressed in the catagen; (4) Cluster four: upregulated in the catagen and downregulated in the other two periods; (5) Cluster five: downregulated in the anagen and upregulated from catagen to telogen; (6) Clusters six and seven: upregulated during the telogen and downregulated in the other two periods; (7) Cluster eight: upregulated during the latter part of the telogen; (8) Cluster nine: downregulated during the catagen. We found the largest number of DEGs in Cluster 6 (upregulated during the telogen), which included 395 genes (17.1%), and the second largest number was in Cluster 2 (downregulated in the telogen) with 380 (16.4%); these results were consistent with the PCA analysis, which also suggested that the developmental transitions from telogen to anagen and catagen to telogen were highly dynamic.
GO and pathway analysis of DEGs
Significant functional categories (p < 0.05) differentially expressed during the yak hair cycle were identified in GO and pathway analysis applied to the DEGs falling into the nine clusters, and 84 GO terms (Additional file 4: Table S2) and 147 KEGG pathways (Additional file 5: Table S3) were enriched with a corrected P-value less than 0.05. The results showed enrichment for particular clusters of gene expression (Figure 4). For example, genes involved in the synthesis of intermediate filament and keratin filament, the Wnt signaling pathway, signaling pathways regulating the pluripotency of stem cells, the Hippo signaling pathway and the MTOR and Hedgehog signaling pathways are greatly enriched among the genes that show peak expression in the anagen and catagen (Clusters C1, C2 and C3). In the process of hair follicle development, the Wnt and Hedgehog signaling pathways are key to stimulation of hair follicle morphogenesis [11, 12], and activation of the pluripotency of stem cells plays an essential role during hair follicle formation [13]. Intermediate filament and keratin filament are essential elements in the process of hair formation [14]. The results presented above suggest that the functions of genes highly expressed in the anagen and catagen are related to signal induction and hair generation. Genes showing peak expression in the telogen (clusters C6 and C7) included some that are required for Natural killer cell mediated cytotoxicity and negative regulation of biological process, the Nature killer cell induces programmed cell death, suggesting that genes that function during the telogen are related mainly to apoptosis.
Signaling and transcription pathways during the hair cycle
After functional analysis, we investigated in more detail changes in the DEGs involved in the biological processes referred to above, in order to provide insights into the relationships among the DEGs during the hair cycle (Figure 5). Some of the DEGs may serve as regulatory signals for proliferation and differentiation of skin and HF cells during development and homeostasis, or play an important part in HF stem cell activation. Differentiation and cycling may involve several key signaling pathways, which include Wnt, Bmp, Shh and Notch. The regulation of mammalian hair cycling is complex, requiring the convergence of many signaling pathways.
Interestingly, we found that Wnt, Notch, and FGF pathway members and the transcription factors that regulate keratin and HF differentiation are more highly expressed in anagen compared to catagen and telogen (Figure 5a). The transition from telogen to anagen occurs when one or two dormant stem cells at the base of the telogen follicle near the dermal papilla are activated by the interplay between Wnt, Shh and Notch factors [15] to produce a new hair shaft [16]. In the Wnt signaling pathway, expression of Wnt family members leads to accumulation of β-catenin mediated through the receptors of frizzled protein, which promote cooperation with TCF/Lef to induce genes encoding factors, such as homeobox gene family members and Shh, that trigger the hair cycle to transition from telogen (resting) to anagen (growth) [17]. Many key transcription factors are involved in the process described above (Figure 5b). BAMBI interacts with the Wnt receptor Fzd and the co-receptor LRP6 and promotes Wnt-induced cell cycle progression and proliferation [18]. CUX1 stimulates TCF/β-catenin transcriptional activity [19]. JAG1 and CDH3, as target genes of the WNT signaling pathway, are key factors in the transition of hair from early anagen to late anagen [20]. HF differentiation is characterized by development of all the compartments of the HF, and different signal molecules play specific roles in this process. FoxN1 and Notch gene products play an important role in regulating HF differentiation with Wnt5a mediators [21]. DLX-3 acts as a transcription factor downstream of Wnt signaling; it controls the expression of keratin in hair development, and along with DSG4 it controls hair follicle cell differentiation [22, 23]. VDR and Hr are two gene products essential for normal hair cycling in mammals [24]; they suppress Wise expression in vivo and allow hair cycle progression [25].
Among the genes highly expressed during catagen, we found a series of transcription factors that regulate the transition of the hair cycle from anagen to catagen. Fgf18 expression in hair follicles is strictly associated with the catagen and telogen phases of the hair growth cycle; it will immediately inhibit the proliferation of stromal cells and strongly suppress the growth of hair follicles [26]. Wise inhibits the Wnt signaling pathways in the course of the hair cycle by binding to LRP receptors [27]. BMP4 can interact with Fgf18 to inhibit the activation of hair follicle stem cells. Fgf5 and its receptor can promote the transition from anagen to catagen [28]. We found that BMP4 and Fgf5 are highly expressed in both anagen and catagen stages; this may indicate that they have a more prolonged effect on the hair cycle than other transcription factors and they may have some unknown effect during anagen.
The telogen can be divided into refractory (early period) and competent (end period) stages [29]. In the early to mid telogen, growth-suppressing signals such as DKK1 are in play, followed by active repression of quiescence mediators, mainly PDGF and BMPs, during late/competent telogen [30].
Co-expression network construction and analysis
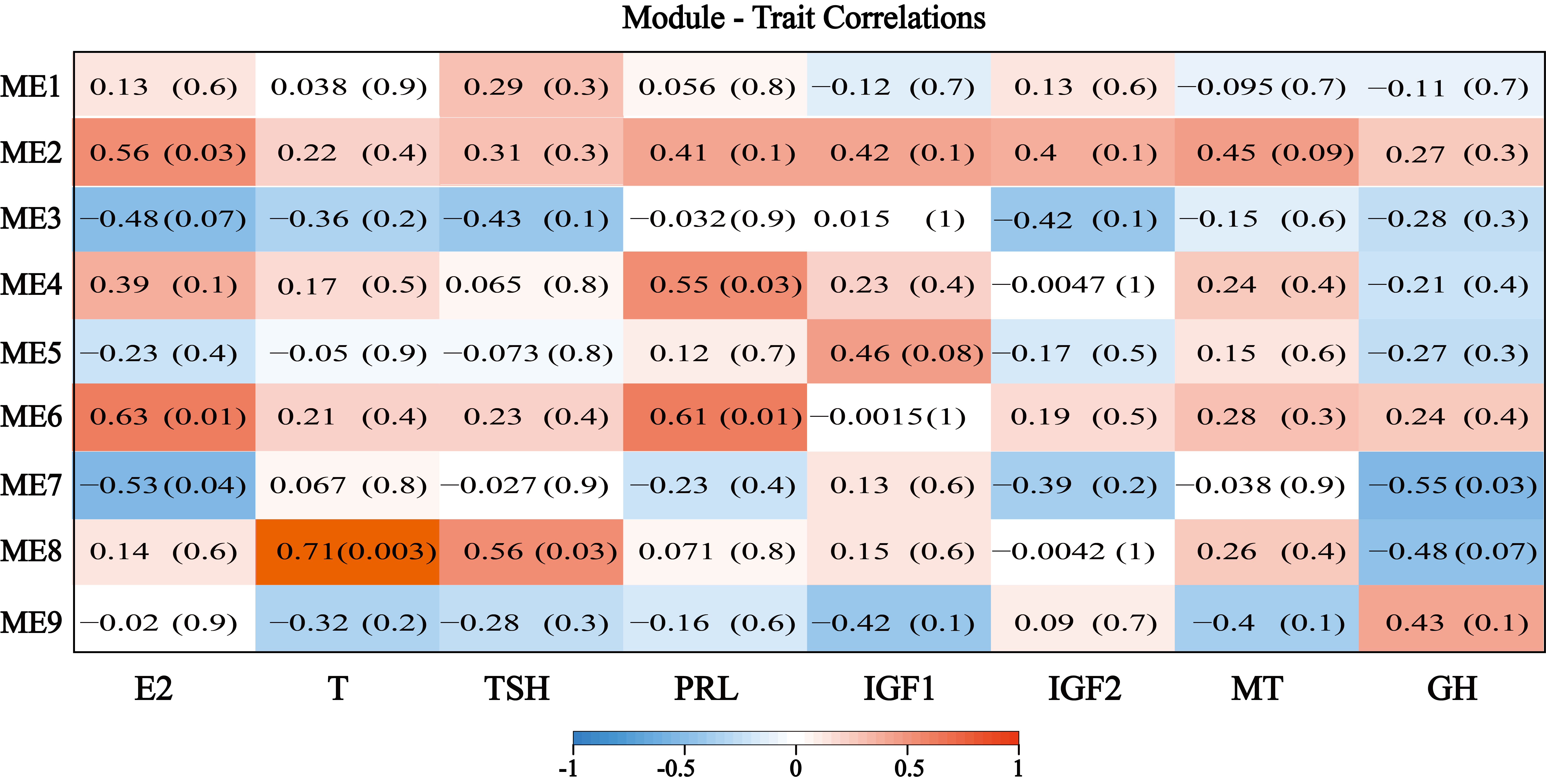
To extract additional biological information embedded in the transcriptome data set, we used the WGCNA software package in R to identify modules of co-expressed genes, and a total of 10 modules were obtained (Figure 7a). Among these 10 modules, the grey module represents the genes that cannot be assigned to any module, and the number of genes contained in each module varies widely. The black module (M3) contains the fewest, only 49 genes, while the blue module (M8) contains the most, with 1,015 genes. We measured the in vivo hormone contents from different samples (Additional file 6: Table 4) and correlated them with the module (Additional file 7: Figure S3). We found that the correlation between module M8 and testosterone content was 0.71 (p value 0.003), indicating that the genes in M8 may be associated with testosterone. ME8 consisted of 1,015 genes with a developmental trend similar to testosterone (T) that showed the highest expression levels in catagen (Figure 7b). In previous studies of humans, T is converted to di-hydro-testosterone (DHT) by 5α-reductase, then binds to the androgen receptor in the hair follicle target cells, inhibiting the growth and development of the dermal papilla, interfering with the growth and metabolism of hair follicle cells, and advancing the hair into the rest period [31]. The genes with the highest degree of connectivity in the module and had high gene significance measures with the hormone are called hub genes and are considered to have important functions within the module (Figure 6c). In the genetic interaction network, we found that ME8 hub genes included genes SLF2, BOP1, DPP8 (Figure 6d), explain that they are the pivotal genes of the module, which may play an important role in the process of testosterone affecting the hair cycle.
{kind=link}
{kind=link}
{kind=link}