Clinical research
Patients with non-traumatic ONFH, who were hospitalized for surgical treatment in Liaocheng People’s Hospital, were randomly selected according to the guidelines for clinical diagnosis and treatment of adult femoral head necrosis (2019) [27]. Patients were diagnosed by 2 clinicians based on X-ray, CT or MRI imaging systems. The necrotic frozen tissues or formalin-fixed and paraffin-embedded (FFPE) sections were derived from hip replacement surgery. After hematoxylin-eosin staining, pathological diagnosis was performed under light microscope. The remaining tissues were frozen in liquid nitrogen within 30 minutes post-operation. Simultaneously, 2 ml of peripheral blood was collected into an EDTA anticoagulation tube and stored at -80°C. In addition, 30 healthy people matched with those patients in terms of age and gender were selected as controls. 2 ml of peripheral blood from a healthy control was collected and stored in a refrigerator at -80°C. According to the Declaration of Helsinki, this study was approved by the Ethics Committee of Liaocheng People's Hospital. Each participant had signed informed consent form prior to sample collection.
32 patients with non-traumatic ONFH included 26 males and 6 females, aged at 59.56±9.517 years. 4 (12.5%), 8 (25%) and 20 patients (62.5%), respectively, suffered from the left, the right, and bilateral femoral head necrosis. The diseased tissue was localized through radiographic, pathological (Figure 1 A-C) and cytological images (Figure 1 D-E).
Whole Exome sequencing (WES)
Firstly, genomic DNA was extracted from whole blood by using QIAamp DNA blood MIDI Kit (Qiagen, Germany). Quality control was performed by using a nanodrop (Thermo Fisher Scientific, USA), with an OD 260/280 ratio of 1.8-2.0 [28].
Secondly, DNA was interrupted into ~200 bp fragments with Covaris S220. Breaking parameters were set up as follows: Duty factor 10%; Peak Incident Power 175; Cycles per Burst 200; Treatment time 360s; and Bath Temperature 4 °C -8 °C. Agilent 2100 quality control was performed on fragmented DNA.
Thirdly, Agilent Sureselect DNA Targeting Sequence Capture Kit was applied for library preparation as follows: (1) End repair was performed on fragmented DNA, where A was added to 3' end, and all gaps were connected with adapters. After each step, AMPure XP beads were used for purification. (2) Polymerase chain reaction (PCR) was performed with an amplification volume of 50 µl. The program was set up as follows: 98°C pre-denaturation for 2 minutes; 98°C denaturation for 30 seconds, 65°C annealing for 30 seconds, 72°C extension for 1 minute, totally 10 cycles; 72°C extension 10 min; and 4°C, hold. The product was purified with AMPure XP beads. (3) Amplified DNA was hybridized and placed at 65°C for 16-24 hours. (4) After hybridization, stranded penicillin magnetic beads were applied for probe capture and PCR amplification, with an amplification volume of 50 µl. The program was set up as follows: pre-denaturation at 98°C for 2 min; denaturation at 98°C for 30 seconds, annealing at 57°C for 30 seconds, and extension at 72°C for 1 min; totally 12 cycles; 72°C extension for 10 minutes; and 4°C, hold. (5) AMPure XP beads were used for purification, whereas Aglinet 2100 for quality control. The fragment size was 250 bp-350 bp, and thus, library preparation was completed.
Finally, Nextseq 500 (Illumina) was applied for PE75 sequencing.
Data quality control and general analysis
Trimmomatic (V 0.36) was used to remove the index and to filter out low-quality bases. BWA software (V 0.7.17) was employed to map the filtered reads with the hg19 human genome as a reference. Picard was applied to mark and delete repeated sequences. GATK4.1.8.1 Recalibration was integrated for local mapping whereas base recalibration for the variants calling program.
Germline mutation analysis
Haplotype Caller was performed to call variants of patients’ and healthy controls’ blood samples. Variant Recalibrator and Apply Recalibration were applied for quality control and filtering (Broad Institute) [29]. ANNOVAR was conducted to annotate each variant to predict potential functional impact [30]. Plink was used for association analysis to determine genes and sites with significant allele frequencies of >0.01, and to count the sites with allele frequencies of ≤ 0.01 in patients’ blood samples.
Somatic mutation analysis
a. Somatic mutation calling in self-matched sample
MuTect2 (Broad Institute) [29] was performed for somatic mutation calling. Firstly, the paired peripheral blood sample data were processed to construct a PoN and to provide filtering support for calling somatic mutations. Secondly, preliminary somatic mutation calling was performed to correct and filter those original mutations. The resulting somatic mutations were annotated by VEP. Variance analysis was performed to predict potential functional impact of each variation [31]. Finally, vcf2maf software was used to convert annotated VCF file into MAF file, whereas maftools software package was applied to visualize variation distribution.
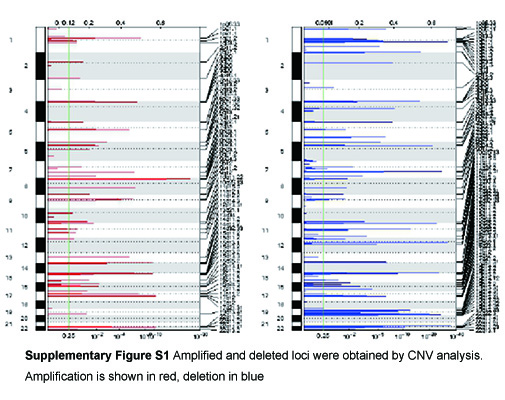
b. Copy number variation (CNV) analysis
Using CNVKit [32], Binary Alignment Map of 32 matched necrotic tissues and blood samples with the hg19 human genome as a reference was established. CNV fragments of 32 samples were obtained and calculated via GISTIC [33], with a q<0.25.
c. Prediction of high-frequency variant genes
MutSigCV [34] (version 1.41) was used to identify somatic SNV and InDel variant and to predict those genes with high-frequency variants, with a q<0.01.
Construction and analysis of PPI network
HTRIdb and HPRD databases were employed to query interaction proteins among those identified genes. STRING [35] was performed to construct a PPI network. Cytoscape v3.7.2 [36] was applied for network visualization. MCODE [37] was conducted to detect closely connected network submodules in a PPI network.
GO and KEGG enrichment analysis
GO (Gene Ontology) functional enrichment analysis and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway annotations were performed on candidate genes, with a p<0.05 as the minimum standard for any significant difference.
{kind=link}