Clinical features of the Rett syndrome patient with FOXG1 mutation
This patient was a 1 year and 11 months old female infant who was referred to our department because of psychomotor developmental delay. The proband was delivered at term to a 34-year-old mother by Cesarean section due to breech position at 2019/12/16. Her birth weight was 3.09 kg and there was no history of asphyxia at birth. Her mother accepted all of the regular inspections as required and no abnormalities was found during her pregnancy. She was the second child of a non-consanguineous couple. Her five-year-old brother had a normal developmental trajectory. The patient was found developmental delay after birth and diagnosed with developmental delay till she was 7 months old.
For physical development, her height was 77cm (<P3) and weight was 10kg (P18), which suggested to short stature. Her occipitofrontal head circumference (OFC) was 42cm (<P3), referred to be microcephalus. For psychomotor development, she could not gain full head control and smooth roll over. She could neither sit nor stand without help. She could not develop hand grasping with specific purpose. Excessive unconscious hand movements were observed such as shaking and flapping. Eye contact was very few. Lack of speech development was observed. Hypotonia was observed after birth. Feeding difficulties were seen after adding semisolid food. Sleeping disturbance has been observed soon after birth and lasted till now. She also had bruxism while awake.
When she was 1 year and 6 months old, seizure was first observed with a sudden loss of consciousness, eye gaze and limb spasms that spontaneously resolved after1-3 minutes. Episode of seizure occurred almost daily. She was diagnosed with generalized tonic-clonic seizures and began to receive antiseizure therapy two months later with oral antiepileptic drugs (Topiramate, Depakin and Topamax and i.v. drip of adrenocorticotropic hormone (ACTH). The seizures were relieved, but not completely controlled. At present, the seizures still occurred 2 or 3 times a month.
Her first regular electroencephalogram (EEG) at seven months’ old was normal. Her 24-hours video EEG at 1 year and 8 months showed generalized temporal spike and spike-slow waves during wakefulness and sleep stage. Her first brain magnetic resonance imaging (MRI) scan at seven months’ old showed widened bilateral temporal extracerebral space (the widest spaces were 14mm at right and 19mm at left, respectively), small bilateral temporal lobes and delayed myelination development (equivalent to 3-4 months). At the age of 1 year and 2 months, she took her second MRI scan. Except widened bilateral temporal extracerebral space, small bilateral temporal lobes and delayed myelination development (equivalent to 6-7 months) as before, corpus callosum hypoplasia was also observed.
She accepted examinations of neural electrophysiological examination involving flash visually evoked potential (fVEP) and auditory brainstem response (ABR) when she was nine months old. The fVEP showed bilateral prolonged baseline P100 and N145 latencies. ABR showed bilateral prolonged latencies of wave I, III and V. No abnormality of urinary system was found by color Doppler ultrasound at 10 months old. DR X ray film for hip joint anteroposterior and abducent projections at 1 year and 11 months old found no abnormalities.
The female child showed obvious neurodevelopmental delay after birth without a period of relatively normal development. She had typical clinical phenotypes involving impaired social interaction, lack of speech development, delayed motor development, stereotypic movements of hands, hypotonia, bruxism while awake, sleep rhythm disorder and seizure. Combined with characteristic brain MRI, the child was diagnosed with Rett syndrome, congenial variant (OMIM#613454) caused by FOXG1 mutations.
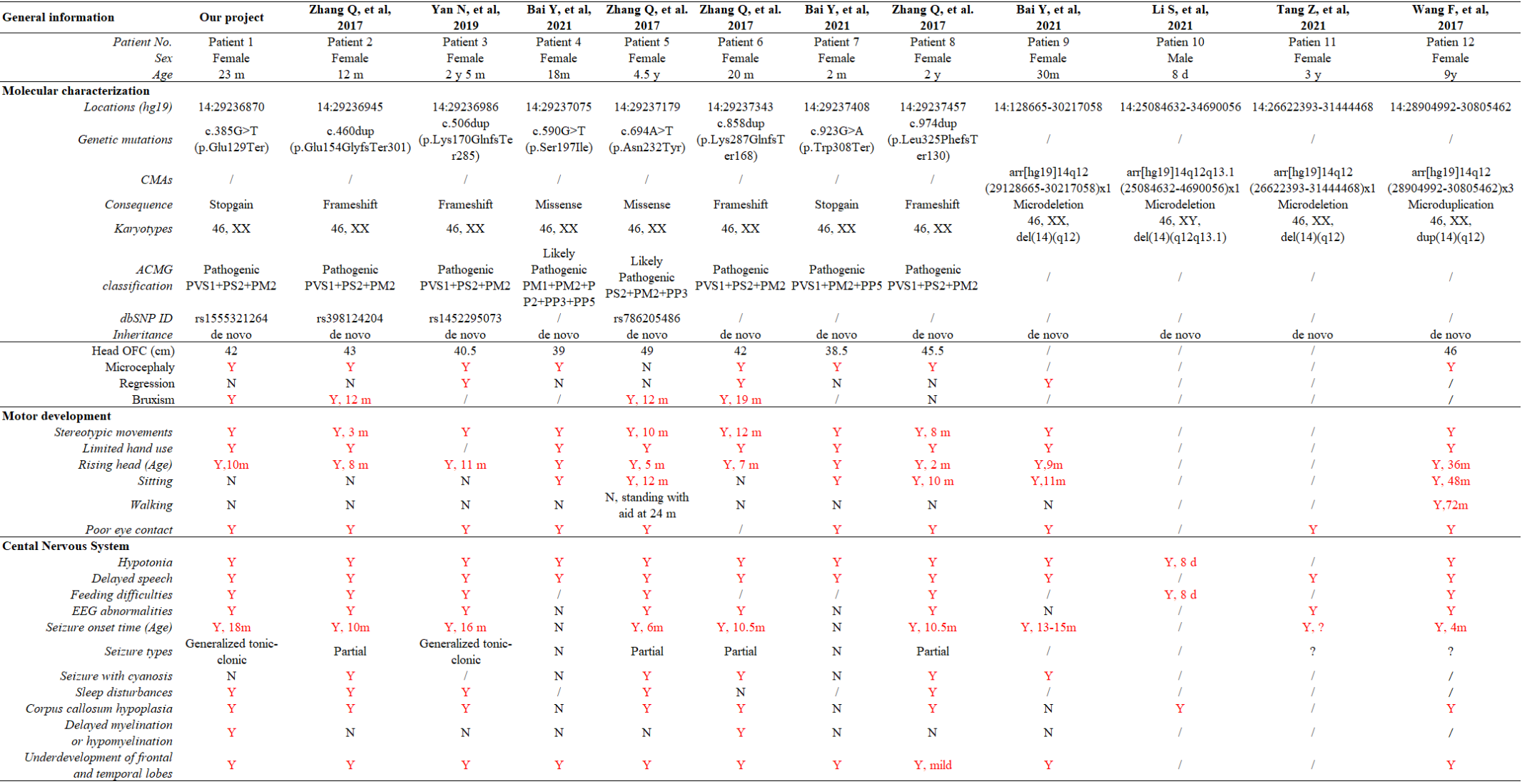
Comprehensive analysis for 12 Chinese with FOXG1 mutations
In the published reports, 11 individual Chinese cases with FOXG1 mutations were compiled. To have a comprehensive re-analysis for these FOXG1 mutations, the case in our sample dataset was put together with the other 8 cases. After carefully reviewing the published articles and inquiring the medical record data, the clinical phenotypes were compiled in Table 1. Pedigrees of 9 patients were depicted at Figure 2. Ranking of the pedigrees were based on the mutation’s location on FOXG1 protein.
After compiling the described clinical features of the 12 patients (Table 1), we noticed that 11 of them were female (91.67%) and only 1 male (8.33%). Since the clinical descriptions for patient 10 and 11 were scarce, they were left out for the subsequent clinical spectrum analysis. The head size development of eight patients (8/10) was lagging behind the standard of the same age and displayed microcephaly phenotype. Regression was reported in three patients with progressive disappearance of acquired language skills. Bruxism was also identified in four patients. As for abnormalities of the musculoskeletal system, hypotonia and stereotypic movements with limited functional hand use were reported in almost all the patients. At early stages, no patients could walk or sit normally; however, with increasing age, six patients could sit without aid after 10 months. Patient 11 could walk without aid at 3 years old. As for the eyes, almost all the patients had poor eye contact, and could not follow moving objects.
As for the features concerning central nervous system (CNS), delayed speech, feeding difficulties, seizure, abnormal EEG and stereotypic movements were reported in most of the patients. As for seizure, the onset time was at or after 4 months, in our patient 1 at 18 months. Two types of seizures were reported in these patients, partial or generalized tonic-clonic. According to the reported MRI/CT results, corpus callosum hypoplasia and underdevelopment of frontal and temporal lobes were seen in most of the cases. Delayed myelination or hypomyelination was reported in only two cases.
Single-nucleotide mutations (SNMs) identified by next-generation sequencing
FOXG1 was mapped to an evolutionarily conserved region (Figure 3A), and contain only one exon (Figure 3B). Currently, there are 8 heterozygous single-nucleotide mutations (SNMs) of FOXG1 identified in Chinese patients with intellectual disability (ID). The details of these SNMs were described in table 1. One of them (c.385G>T, rs155521264, patient 1) was a stop gain mutation (Figure 3C-D). It caused the codon at position 129 (GAG) for Glutamic acid (Glu, E) to be a premature stop codon (TAG, X) (p.Glu129Ter) (Figure 3-E). As for mutations in patient 2, 4, 5 and 6, all the four mutations were insertions with one nucleotide, which caused frame shifting of the original coding sequence (CDS) of FOXG1 (Figure 3C-E). c.460dup (rs398124204, patient 2) was reported more than 10 times in European patients 22, 25-33 and only once in Chinese 12. c.506dup (rs1450095073, patient 3) and the other two frameshift mutations (c.858dup in patent 5 and c.974dup in patient 6) had been submitted to the NCBI ClinVar database by GeneDx group and reported in Chinese patients with Rett syndrome 12, 34. Although the duplicated locations of the SNMs were different, the CDSs containing these four frameshifting mutations terminated at the same premature stop codon to produce a truncated protein with 453 amino acids (Figure 3F). According to criteria of ACMG classification, these mutations were annotated as “Pathogenic” (PVS1+PS2+PM2).
As for the two missense mutations (c.590G>T, p.Ser197Ile in patient 4and c.694A>T, p.Asn232Tyr in patient 5), they were located in the DNA-binding forkhead domain (FHD). The amino acids (Ser197 and Asn232) were strongly conserved (Figure 3-D) during evolution and not detected in the several public genomic databases, such as 1000 Genome (n = 2,504), NHLBI GO-ESP (n = 6,503), ExAC (n = 60,706), gnomAD (n = 15,708) and TOPMED (n = 60,000). According to the criteria of ACMG classification, they were annotated as “likely pathogenic” (Table 1). For c.590G>T (p.Ser197Ile), it has been submitted to ClinVar by the Genetic Services Laboratory of University of Chicago (Accession: SCV002069287.1) and identified in a patient of 47 months old 35. Analyzed by Missense3D, the Ile substitution disrupts all side-chain / main-chain H-bonds formed by the buried Ser residue (RSA 2.3%) (CO197-Ser OG/CO194-Ile O, CO197-Ser OG/CO201-Arg O; CO200-Lys NZ/CO197Ser O, CO200Lys N/CO197Ser OG, CO201-Arg N/CO197Ser OG). This mutation also results in a switch from the buried Ser (RSA 2.3%) to exposed Ile (26.6%) (Figure 3G-H). The buried H-bond breakage and buried / exposed switch might disrupt the local structure of PHD domain. For c.694A>T (p.Asn232Tyr), it has been submitted to ClinVar database by the DGU-KFSHRC (Developmental Genetics Unit, King Faisal Specialist Hospital & Research Centre) and reported in a Chinese RTT patient 36. The side chain of Asn (N) is a small sized aminocarbonyl, but a bulky p-hydroxyphenyl for Tyr (Y). Analyzed by Missense3D, this mutation led to a switch from a buried Asn (RSA 7.0%) to an exposed Tyr (63.4%), which disrupted all side-chain / side-chain H-bonds (AO187-Asn ND2/AO232-Asn OD1; AO232-Asn ND2/AO231-His NE2; AO236-Asn ND2/AO232-Asn OD1) and two side-chain/main-chain H-bonds (AO232-Asn ND2/AO228-Ser O and AO236-Asn ND2/AO232-Asn OD1). The Tyr232 only forms one side-chain/main-chain H-bond with Asn236 (AO236-Asn ND2/AO232-Tyr O) (Figure 3I-J).
To comprehensively understand the mutation patterns of FOXG1, variants annotated as “Pathogenic” and “Likely Pathogenic” from NCBI ClinVar and DECIPTHER were aligned against the CDS of FOXG1 (Supplementary Figure S2). Totally, 171 mutations in the CDS of FOXG1 were recruited in both databases. 38.60% (66/171) were missense mutations, 37.43% (64/171) frameshift, and 23.98% (41/171) nonsense. For the missense mutations, about 93.94% (62/66) were in the forkhead domain (FHD) which was responsible for DNA binding, 4.55% (3/66) in the JARID1B binding domain (JBD) responsible for the interaction between FOXG1 and JARID1B (also called as KDM5B). Only one likely-pathogenic missense (c.1439A>G, p.Gln480Arg) was localized in the C-terminal disordered region. As for the pathogenic insertion/deletions (ins/del), they were distributed throughout the whole region of FOXG1.
Copy number variations (CNVs) containing FOXG1 gene
Through oligonucleotide array-CGH, CNVs were identified three microdeletions in patient 9, 10, 11 and one microduplication in patient 12 (Table 1). As for the 3 microdeletions, Li.S_2021 was about 9.61 Mb in length (14:25,084,632-34,690,056) and contained 19 protein-encoding genes, including FOXG1 37. As for Tang.Z_2021, it was completely covered by Li.S_2021. This deleted region was about 4.82 Mb in length (14:26,622,393-31,444,468), and contained 7 protein coding genes (Table 2) (NOVA1, FOXG1, PRKD1, G2E3, SCFD1, COCH and STRN3). For Bai.Y_2021, it was the shortest microdeletion (14:29128665-30217058, 1.09Mb) and contained only two genes, FOXG1 and PRKD1 38. A 1.90 Mb microduplication at 14q12 (14:27974743-29875213) was identified in patient 12 and contained only the FOXG1 gene (Supplementary Figure S3). 14. There were no chromosomal aberrations detected in her parents.
Table 2. Haploinsufficiency predictions for the 7 shared genes in microdeletions of patient 8 and 9
|
Name
|
pHI
|
Inheritance
model (pAD)
|
Phenotype / OMIM
|
Development Disorder
Genotype – Phenotype
(DDG2P)
|
Gene Curation
Coalition (GenCC)
|
ClinGen
|
|
NOVA1
|
2.330
|
0.977: VLD
|
/
|
/
|
/
|
/
|
|
FOXG1
|
3.110
|
0.998: VLD
|
613454 /Rett syndrome, congenital variant
|
·Monoallelic;
·Loss of function;
·Congenital Variant of Rett Syndrome;
|
·Definitive: 3
·Strong: 1
|
·Definitive: AD
·Haploinsufficiency: 3
·Triplosensitivity: 0
|
|
PRKD1
|
2.790
|
0.698: LD
|
617364 / Congenital heart defects and ectodermal dysplasia (CHDED)
|
·Monoallelic;
·All missense/in frame;
·Syndromic Congenital Heart Defects
|
Limited: 1
|
/
|
|
G2E3
|
13.460
|
0.213: LR
|
/
|
/
|
/
|
/
|
|
SCFD1
|
9.330
|
0.822: VLD
|
/
|
/
|
/
|
/
|
|
COCH
|
15.090
|
0.355: LR
|
·601369 / Deafness, autosomal dominant 9 (DFNA9)
·618094 / Deafness, autosomal recessive 110 (DFNB110)
|
/
|
Definitive: 1
|
Definitive: AD
|
|
STRN3
|
7.730
|
0.725: LD
|
/
|
/
|
/
|
/
|
Notes: VLD: Very likely dominant; LD: Likely dominant; VLR: Very likely recessive; LR: Likely recessive. pAD: probability of being autosomal dominant
There were 177 individual deletions in the genomic region (Li.S_2021, chr14: 25,084,632-34,690,056) recruited in different public databases, such as 10 in ClinGen (Clinical Genome Resource), 101 in NCBI’s ClinVar and 66 in DECIPHER (Figure 4C- E) databases. Out of these microdeletions, 40 of them (22.60%) spanned the CDS of FOXG1 gene: 6 in ClinGen, 12 in ClinVar and 22 in DECIPHER databases. Six short deletions just contained the FOXG1 gene, in which the three shortest deletions, 1057648 (chr14: 29,236,486-29,237,955, 1.47Kb, ClinVar), 820618 (chr14: 29,236,466-29,237,975, 1.51Kb, ClinVar) and 290100 (chr14: 29,236,278-29,237,804, 1.52Kb, DECIPHER) just covered the protein-coding sequence of FOXG1.
Mutation analysis of PRKD1 gene
PRKD1 was also contained in the two microdeletions of patient 8 and 9. Therefore, it’s necessary to assess the contribution of PRKD1 to the clinical phenotype of FOXG1-related RTT disorder. After compiling the pathogenic mutations in the CDS of PRKD1 from published articles, ClinVar and DECIPHER databases, thirteen patients were found to have single nucleotide mutations (SNMs) annotated as pathogenic or likely pathogenic (Figure 5A and Supplementary Table S1). Based on the mode of inheritance of these patients, five were heterozygous (4 de novo and 1 unknown), 7 homozygous (from two consanguineous families) and 1 unknown. It’s worth noting that for patients with homozygous mutations, all of them suffered only from non-syndromic congenital heart diseases (CHDs), no other systemic phenotypes were manifested 39, 40. The heterozygous mutations could lead to not only CHDs, but also abnormalities of the CNS, such as intellectual disability, global developmental delay, hearing impairment, delayed language development, or microcephaly 41, 42.
There were also 5 microduplications and 9 microdeletions completely or partially covering only PRKD1 recruited in ClinVar and DECIPHER databases (Figure 5B, Supplementary Table S2). Since there were no clinical phenotypes for 4 microduplications (14q12, nssv13647033, nssv13643620 in ClinVar, and 380299 in DECIPHER), 10 CNVs (9 microdeletions and 1 microduplication) were remained for subsequent analysis. Except for abnormalities of cardiovascular system (Tetralogy of Fallot, Ventricular septal defect, hypertension) in 6.45% (4/62) of all the reported clinical phenotypes, there also exhibited RTT-like phenotypes, such as facial dysmorphisms (17.74%, 11/62), intellectual disability (11.29%, 7/62), microcephaly (8.06%,5/62), hypotonia (6.45%, 4/62), seizure (4.84%, 3/62), hypoplasia of the corpus callosum (3.23%, 2/62), delayed language development (3.23%, 2/62), behavioral abnormality (3.23%, 2/62), feeding difficulties (3.23%, 2/62), global developmental delay (1.61%, 1/62), agenesis of corpus callosum (1.61%, 1/62), delayed myelination (1.61%, 1/62), simplified gyral pattern (1.61%, 1/62) and others (11.29%, 7/62) (Figure 5C). As for others, it included bruxism, hypothyroidism, intrauterine growth retardation, microdontia, nystagmus, obesity and short stature. It’s very likely that serine/threonine-protein kinase D1 (PRKD1) acts an independent contributor or collaborator with FOXG1, for the clinical phenotypes of congenital variant form of Rett syndrome.
Intergenic regulatory elements in the region between FOXG1 and PRKD1
According to reports about large-scale identification of functional elements in the human genome revealed by the Encyclopedia of DNA Elements (ENCODE) Consortium, intergenic noncoding regions often contain multiple regulatory elements such as enhancers, silencers or insulators 43. There were many CNVs covered the intergenic genomic region between FOXG1 and PRKD1 (FOXG1-PRKD1). To evaluate the contribution of intergenic CNVs to the phenotypes of FOXG1-related RTT disorder, longer CNVs spanning the whole region of FOXG1 plus PRKD1 were removed and only those partially covering FOXG1 or PRKD1 were left for subsequent analysis (Figure 6A-B).
Genomic sequences from zebrafish, Xenopus tropicalis, chicken, opossum, rat, mouse, dog, Rhesus macaque and chimpanzee were compared against human genome (chr14:28,827,675-30,608,124, hg19), FOXG1 was strongly conserved during evolution in all the selected animals. As for PRKD1, it was appeared in mammals and might be a mammalian-specific gene (Figure 6C). Besides, eight strongly-evolutionarily conserved regions (SECRs) were identified in the intergenic FOXG1-PRKD1 region (Figure 6-C). According to the chromatin modification patterns from ENCODE project, the FOXG1-PRKD1 region might contain regulatory elements, such as enhancers or silencers (Figure 6D). According to, fourteen annotated regulatory elements were contained in this genomic region, which were collected in the Open Regulatory Annotation (version 3.0, ORegAnno) database 44 (Figure 6E). Most of them overlapped with the SECRs. For the annotated regulatory elements, 12 of them were tested for the enhancer activity in transgenic mouse embryos (E11.5) with LacZ staining in VISTA enhancer browser 45. Six of the SECRs acted as active enhancers, 3 in mouse forebrain (hs566, hs1523 and hs342), 2 in hindbrain (hs1539 and hs1168) and 1 in neural tube (hs598) (Figure 6F-G). After analyzing the in situ Hi-C (High-through Chromosome Conformation Capture) data for seven human cell lines (GM12878, K562, KBM7, HMEC, HUVEC, IMR90 and NHEK), this region has three topologically associating domains (TADs). FOXG1 and the intergenic region (FOXG1-PRKD1) were completely constrained in the second TADs, which was completely encompassed by linkage disequilibrium (LD) blocks which were divided by recombination hotspots (Figure 6H-I).
{kind=link}