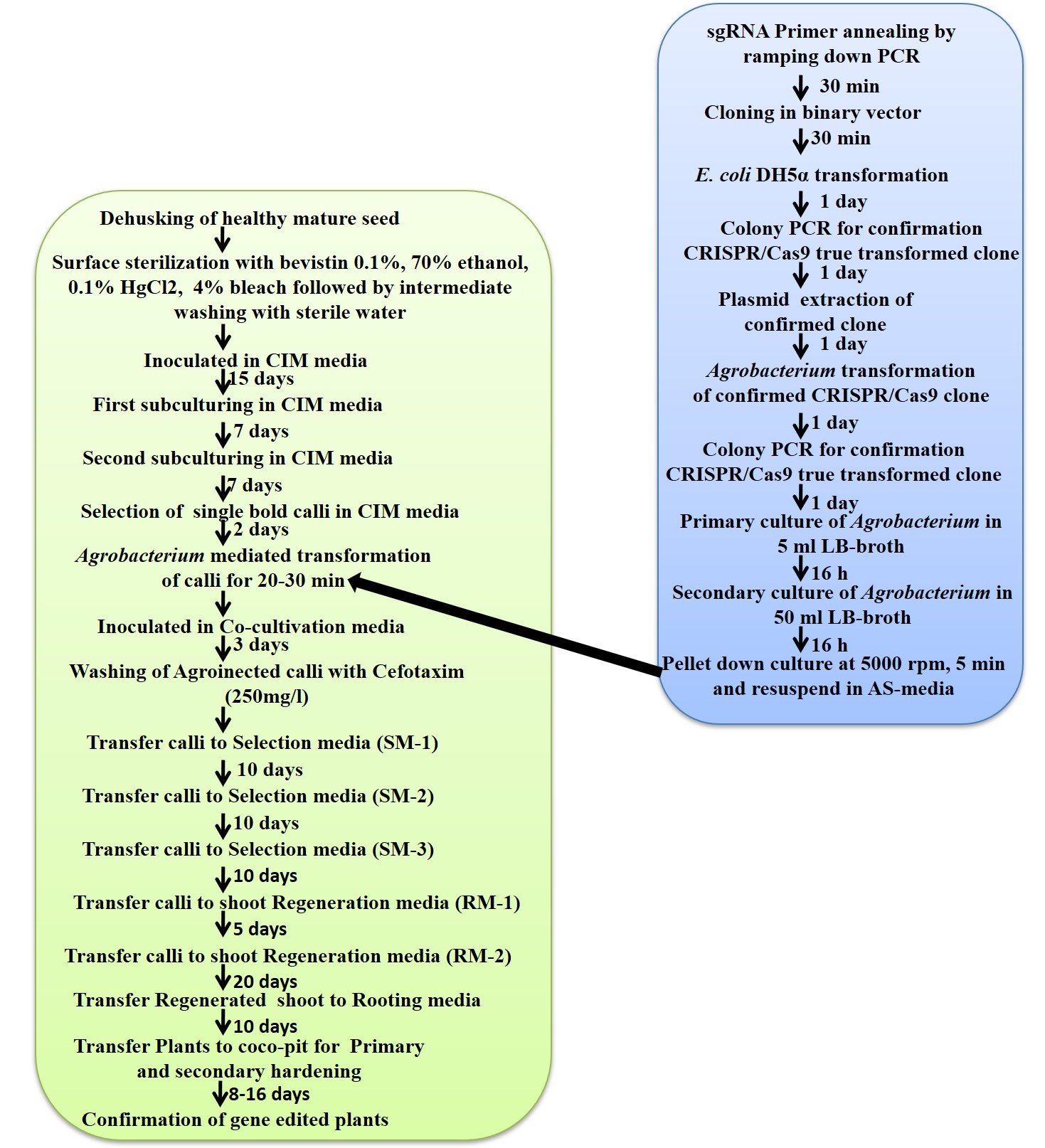
Primer Designing: There are various tools available for designing gRNA in the plant [14]. In this study, CHOPCHOP software was used to design gRNA from the target gene sequence (query sequence) using default parameters. The CHOPCHOP software analyses the query sequence and displays all possible 20-bp sequences identified immediately after the PAM (NGG). Parameters, including length of gRNA, off-targets, etc., may be changed as per our requirements. The sequence showing zero self-complementarity and optimum GC content was used for designing gRNA. Further, the T/G sequence from the 5' end and the AGG (or) CGG sequence from the 3' end of the selected gRNA primer were removed. BsaI restriction enzyme site (GGCG) was added at the 5' end of the forward primer (Fp) and AAAC at the 3' end of the reverse primer (Rp) (Fig.1).
Primer annealing of gRNA: For primer annealing, 1µl each of forward (Fp) and reverse (Rp) primers from 100 µM of stock solution of primers was added to 8 µl nuclease-free water. Primer annealing was performed by ramping down the PCR method. The PCR programme was set as follows: hot start at 95 ºC, 2 min, start cycle 70 times, temperature step (processing temp: 95 ºC, 10 sec, temp. increment: -1), end cycle 70 times, storage at 4 ºC, 15 min. After completion of the primer annealing step, PCR tubes were immediately kept on ice, followed by the addition of 90 µl of nuclease-free water to 10 µl of a primer reaction system for further use (Fig. 2).
Cloning and ligation of gRNA: Annealed gRNA was cloned into the pBUN411 binary vector using the NEB Golden Gate Assembly Kit (BsaI-HFv-2) (E1601S) by following the manufacturer's protocol (Fig. 2).
Transformation of E. coli with recombinant plasmid DNA: The ligated mixture was added to a 50 µl aliquot of competent cells (E. coli DH5α), mixed by gentle tapping, and incubated on ice for 30 min. Heat shock was given at 42 oC for 1 min, kept on ice for 1 min, and then 950 µl of sterile LB broth was added to it. The tube was incubated at 37 oC in a rotary shaker (200 rpm) for 1 h. A hundred microliters of cell suspension were spread on LB agar containing kanamycin (50 mg/ml) for the selection of transformants (Fig. 3).
Colony PCR: Colony PCR was performed to screen recombinant plasmids using vector and gRNA-specific primers (OsU3_Fp and gRNA_Rp). The amplified PCR product was electrophoresed on a 1% agarose gel. The presence of a 300 bp band indicates the recombinant colony.
Isolation of plasmid DNA: The recombinant colony was inoculated in 5 ml of LB broth containing kanamycin (50 mg/ml) and grown for 10–12 h at 37 ºC in a shaker. A part of the overnight grown culture (1.5 ml) was centrifuged in a microfuge tube and the bacterial pellet was resuspended in 100 μl of solution I (25 mM Tris pH 8.0, 10 mM EDTA pH 8.0, 50 mM Glucose). Immediately, 200 μl of freshly prepared solution II (0.2 M NaOH, 1% SDS) was added to the tube and kept on ice for 15 minutes, mixed by inverting the tubes. The ice-cold 150 μl of solution III (5 M potassium acetate: 60 ml, Glacial Acetic Acid: 11.5 ml, dH2O: 28.5 ml) was added, gently mixed, and centrifuge at 12000 rpm for 10 minutes. The supernatant was collected, add 300 μl of chloroform: isoamyl alcohol (24:1), was added and mixed well by inverting the tube several times, followed by centrifugation at 12000 rpm for 5 min. The aqueous phase was transferred into a fresh microcentrifuge tube and 300μl of isopropanol was added and mixed gently by keeping it at RT for 2 min, followed by centrifugation at 12000 rpm for 10 min, then discard the supernatant. The DNA pellet was obtained by centrifugation at 12000 rpm for 10 min, washed with 70% ethanol, and re-suspended in 50 μl of TE (10 mM Tris pH 8.0, 1 mM EDTA pH 8.0). The yield of plasmid DNA obtained was around 5-7 μg.
Transformation of A. tumefaciens with recombinant plasmid DNA: Competent cells of the A. tumefaciens AGL1 (pSoup) strain were used for transformation with plasmid DNA. The competent cells were prepared as follows: A single colony of the A. tumefaciens strain was inoculated in 3 ml of LB broth containing rifampicin (50 mg/ml), tetracycline (5 mg/ml), and incubated at 28 °C over-night in a rotary shaker (200 rpm). Fifty milliliters of fresh LB broth were inoculated with 1 ml of grown culture and incubated at 28 ºC for 4-5 h till the O.D. (A600 nm) reached around 0.6-1 and chilled on ice. The cells were pelleted down by centrifugation at 5000 rpm for 10 min at 4 ºC, and re-suspended in 20 ml of ice-cold sterile water. After centrifugation for 5 minutes, the pellets were resuspended in 20 ml of ice-cold sterile water and incubated at room temperature for 5 minutes. The cells were pelleted again and resuspended in 1 ml of 20% glycerol before being aliquoted (200 μl) into 1.5 ml of an Eppendorf tube. The aliquoted cells were frozen quickly in liquid nitrogen and then stored at -80 ºC. The recombinant clones were mobilised into A. tumefaciens competent cells using the freeze-thaw method. The transformed A. tumefaciens colonies were confirmed by colony PCR using primer pBUN411 vector OsU3_Fp + gRNA_Rp (Fig. 4).
Callus induction: Dehusked mature seeds of Oryza sativa cv. Rajendra Kasturi was used for callus induction as described previously [15]. In brief, dehusked seeds were washed twice with sterile distilled water followed by surface sterilisation with bavistin 0.1% for 20 min with intermittent shaking, followed by washing three times with sterile distilled water. Seeds were further treated with 70% ethanol (v/v) for 30 sec, followed by washing thrice with sterile distilled water. Seeds were then treated with 0.1% HgCl2 for 5 min with intermittent shaking, followed by washing thrice with sterile distilled water and air-drying on sterile filter paper for 5 min. The air-dried seeds were placed on a callus induction medium (CIM) (Fig. 5) and incubated for 15 days at 28 ºC in the dark (Fig. 6A).
Callus subculturing: For the first subculturing, the seed portion and shoot portion of embryogenic calli were removed with the help of forceps. Embryogenic calli were broken into medium-sized calli with the help of forceps and kept on new CIM (Fig. 5) and then incubated for 7 days at 28 ºC in the dark (Fig. 6B). For second subculturing, embryogenic calli were broken into small calli with the help of forceps and transferred to new CIM media (Fig. 5, 6B). For genetic transformation, friable calli without any hairy structure were subcultured on new CIM media and incubated at 28 ºC in the dark for 2 days.
Co-cultivation: A. tumefaciens with CRISPR-Cas9 gene construct was grown on LB-agar plate containing rifampicin (50 mg/ml), tetracycline (5 mg/ml), and kanamycin (50 mg/ml), incubated at 28 °C for 48 h, day before transformation process. A single colony of transformed A. tumefaciens harboring desired gene construct was inoculated in 3 ml of LB broth containing an appropriate concentration of rifampicin, tetracycline, and Kanamycin and incubated at 28 °C over-night in a shaker (200 rpm). One ml of fresh culture was inoculated in 50 ml of LB liquid medium containing appropriate antibiotics and incubated overnight at 28 °C in a rotary shaker (200 rpm) followed by pellet down of bacterial culture in next day. The resulting pellet was dissolved in 10–15 ml (depending on the pellet size) of AA-liquid MS media containing 100 μM acetosyringone (AS) (HiMedia Laboratories) (Fig. 5). Scutellum-derived calli were subculture to new CIM media for two days and used for Agrobacterium-mediated transformation (Fig. 6D). The calli were transferred to fresh plastic petridish using a spatula and immersed into AA-AS media (Fig. 5) containing Agrobacterium pellet and swirled intermittently for 30 min. Approximately 1 ml of liquid AA-AS medium was added on top of the co-cultivation medium (CCM) above which a filter paper (Whatman paper No.3) was placed. The transformed calli were taken out from the AA-AS medium and blotted on a sterilized filter paper to remove excess liquid. Transformed Calli were individually placed on filter paper kept above the CCM and incubated in dark for 72 h at 28 °C (Fig. 6E).
Washing of Transformed calli and Selection: The incubated transformed calli were transferred to a sterile 50 ml flask after 72 h of co-cultivation and washed ten times with autoclave sterile water for 1 min followed by 4 times washing with sterile water containing 250μg/l cefotaxime by hand swirling for 15 min then allow to dry on sterile Whatman paper No.3. Thereafter calli were transferred to a selection medium containing phosphinothricin (10 mg/l) (Sigma-Aldrich) (Fig. 5) and incubated in dark at 28 °C for 10 days. Three rounds of selections were done and resistant calli were further preferred for regeneration (Fig. 6F-H). In SM-1 medium (Fig. 5), some of the calli start turning brown and the rest remained creamish colour after 10 days (Fig. 6F) and shifted to fresh SM-2 medium for the next ten days where small soft microcalli started growing from the mother calli (Fig. 6G). Soft microcalli were separated gently and transferred to a fresh SM-3 medium for the next ten days (Fig. 6H).
Shoot and Root Regeneration: After 3 rounds of selection, microcalli started proliferation which was then shifted to the first regeneration medium (RM-1) containing Phosphinothricin (10 mg/l) and cefotaxime (250μg/l) (Fig. 5) and kept in dark for 5 days 28 ºC. In this step calli that were developed into somatic embryos were shifted to RM-2 (Fig. 6I) and placed under photoperiod 16 h light/8 h dark where "green-spot" appeared on regenerated somatic embryogenic calli which further developed into the shoot (Fig. 6I). Shifting of microcalli to light from the dark was necessary because continuous dark conditions resulted in the development of albino plantlets and five days’ dark condition was essential for better regeneration (Prasad et al., 2016). After obtaining a large number of regenerated shoots (>2 cm long), these were subsequently shifted to the rooting medium (Fig. 5, 6J). Root development with lateral took 5-6 days after incubation. All the important steps along with duration were summarized in the form of a flow chart (Supplementary fig. 1).
Hardening: The fully developed plants along with roots were taken out from the culture condition and roots washed smoothly to remove attached trace of media. These plants were immediately transferred to the small plastic pot containing coco-pit and 10 ml half MS-media (Fig. 5K). Afterward, a small amount of coco-pit followed by 5ml distil water was added to the plastic pot and pour 5ml distil water, and immediately covered using a transparent perforated polythene bag. This process would be preferably carried out in evening time. Therefore, the plants were kept in the dusky light of the culture room to stimulate such conditions. The next day plants were kept in proper photoperiod 16 h light and 8 h dark for seven days to serve primary hardening. Then plants were shifted to the greenhouse for secondary hardening and cut the above portion of polybag for proper hardening. Plants were nourished with half MS solution and distilled water on an alternate day. Twenty days after secondary hardening, plants along with coco-pit were transferred to the earthen pot containing coco-pit and soil in a 1:1 ratio (Fig. 6L).
Genomic DNA extraction and PCR analysis: The leaf tissues were harvested and quickly frozen in liquid nitrogen. The genomic DNA was extracted with the modified CTAB method (Doyle and Doyle, 1990). Gene-specific PCR amplification from genomic DNA of WT and gene-edited lines was performed using sets of primer for CaMV35S promoter, OsU3 promoter, PAT gene (Phosphinothricin), and Cas9 gene in a reaction volume of 10μl containing the template genomic DNA (100 ng). PCR tubes were incubated in PCR, steps: initial denaturation step of 94 °C for 5 min, followed by 30 cycles of denaturation (1 min at 94 °C), annealing (the 30s at 52 °C), and extension (40s at 72 °C). After the last cycle, a final extension was carried out for 5 min at 72 °C. The PCR product was electrophoresed on 1.5 % agarose gel (Fig. 6).
Troubleshooting
Even though the method is quite simple, easy, and reproducible but certain things would be taken into consideration.
Devoid of callus induction
- Freshly harvested mature seed lots were used for callus induction to achieve maximum germination.
- Prolonged surface sterilization was avoided as it may cause germination losses.
- Freshly prepared 2, 4-D was used for maximum callus induction.
- Incubation was done at 28 ºC for profuse callus induction.
Agrobacterium culture growth for Co-cultivation and selection
- Inoculate a freshly grown single colony of Agrobacterium harboring a gene construct into 5 ml of LB media containing antibiotics, from this culture 1 ml is used for further incubation. Incubate the inoculation tube at 28 ºC (150 rpm) overnight.
- Agrobacterium harboring gene-culture was transferred into 50 ml of LB broth containing antibiotics Rifampicin (50 mg/ml), Tetracycline (5 mg/ml), and Kanamycin (50 mg/ml).
- The tetracycline concentration was not more than 5 mg/ml.
- Agrobacterium culture O.D. A600 was maintained in the range of 0.8-1.0 in the LB-broth medium. Never preferred the culture OD above or below the range.
- The duration of incubation of co-cultivated calli would not exceed 72 h because more incubation results in the death of transformed calli and Agrobacterium-contamination comes into some of the calli in SM-1 plates after 5 days of incubation.
- In the co-cultivation medium, place properly autoclaved filter paper to avoid excess Agrobacterium.
Low efficiency in selection media
- The fragile embryogenic calli were used for transformation.
- Acetosyringone should never be used if it is more than 48 hours old.
- Co-cultivated calli were washed with sterile (minimum 10 times) containing antibiotic cefotaxime (250 mg/l) after 2-3 days of co-cultivation.
- Freshly prepared cefotaxime (250 mg/l) was preferred. If the color of the cefotaxime solution turns yellow, then it was not used.
- A proper concentration of antibiotics was used in the selection media.
- Added antibiotics to the culture medium when the temperature of the media reaches 50°C.
- Air-dry the washed calli on sterile filter paper for at least 10 minutes in a laminar hood.
Low regeneration frequency:
- Proper concentrations of gelling agents and phytohormones were used in the regeneration medium.
- Freshly prepared phytohormones were used for regeneration media.
- The temperature of the regeneration room (26 ± 2°C) during the culture conditions did not go beyond the mentioned temperature.
- The maximum number of 6-8 green spotted regenerated calli were placed on a single plate.
{kind=link}