In this study, we compared two commercially available DNA extraction methods used in microbiome research: the AllPrep DNA/RNA Mini Kit (APK) and the QIAamp Fast DNA Stool Mini Kit (FSK). Using shotgun metagenomic sequencing, we assessed differences in DNA yield and quality and taxonomic composition while trying to limit the effects of other sources of heterogeneity. We also evaluated whether associations with several phenotypes were affected by the DNA extraction protocol.
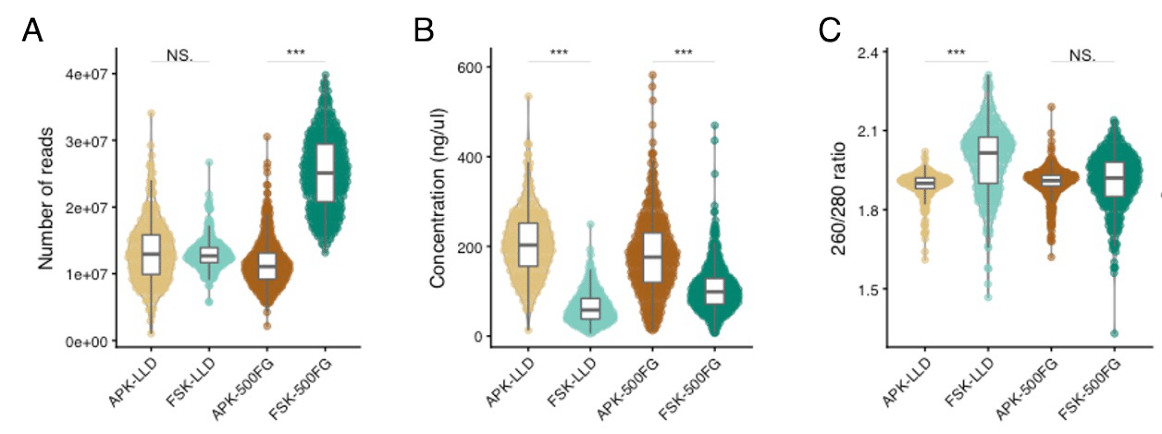
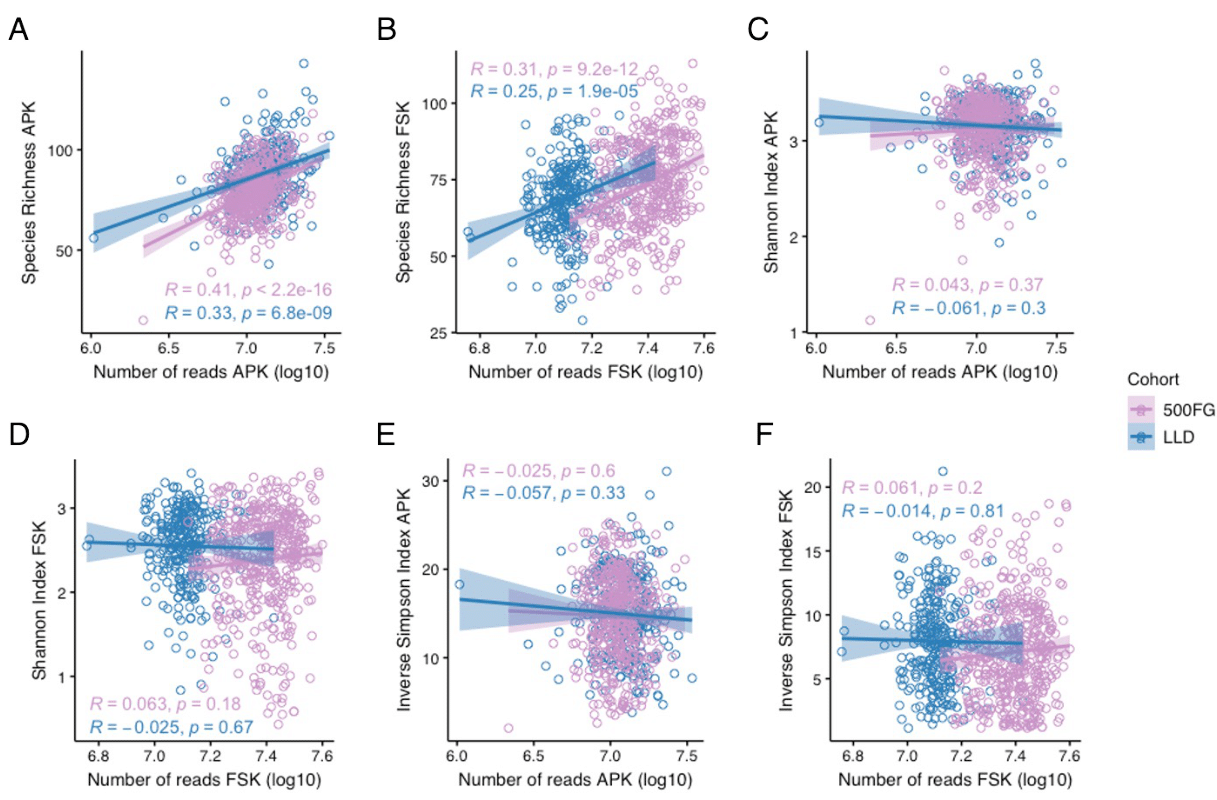
The APK extraction method yielded a higher DNA concentration than FSK. Although only a few studies have compared these two protocols, this difference can be explained by the inclusion of a bead-beating step in the APK procedure. This mechanical disruption has been shown to improve DNA extraction efficiency, independent of the commercial kit used [9]. Although previous studies have described that use of a heating step in combination with the enzymatic lysis used in FSK can also favor bacterial cell lysis by denaturizing the membrane proteins [21], our findings suggest inefficient bacterial DNA recovery with this procedure. While automatization of the FSK protocol could have also contributed to DNA yield dissimilarities, prior research has not found significant differences in nucleic acid concentration and quality between automated and manual methods [12]. Additionally, our results show that the increased DNA concentration in samples extracted with APK resulted in higher microbial diversity and species richness. These findings further support earlier evidence that suggested a higher DNA yield and species diversity in bead-beaten samples [9, 11, 12], pinpointing alpha diversity as an appropriate benchmark for the DNA extraction performance. These results, however, could only be validated in the LLD cohort due to technical variability in the DNA concentration measurements in 500FG. In addition, we did not find a significant correlation between alpha diversity indices and read depth, discarding a potential impact of read depth differences on observed diversity values.
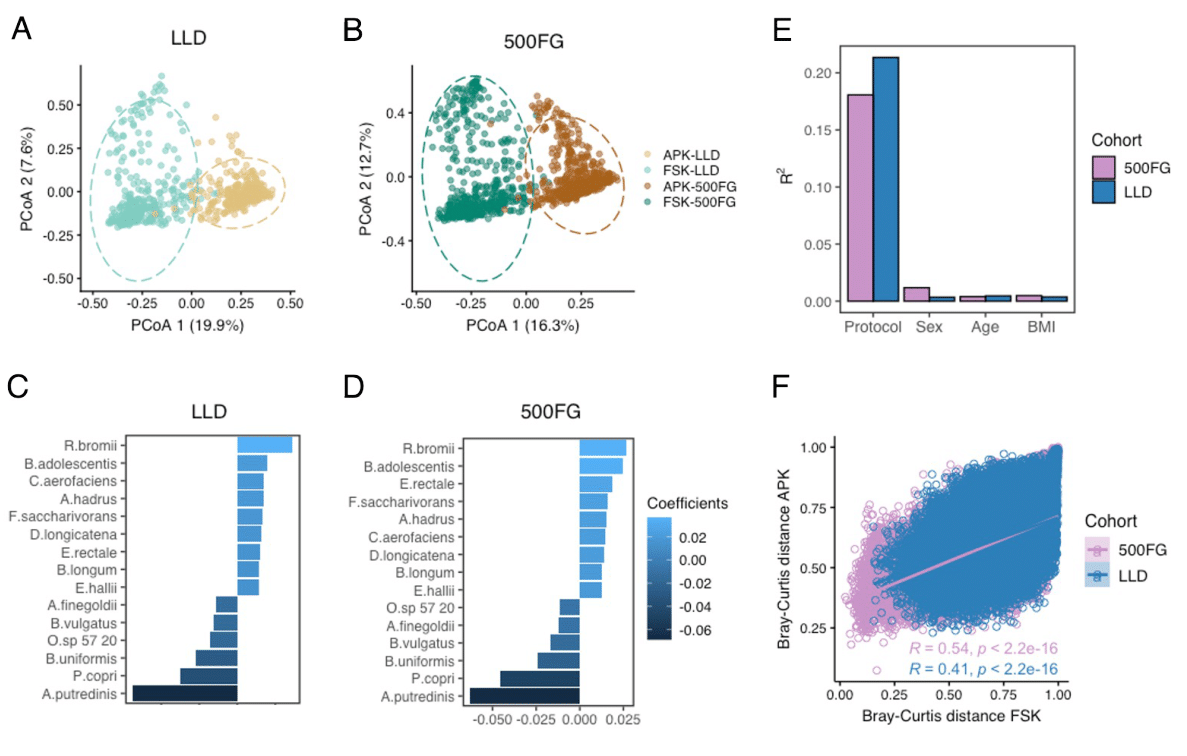
Our analysis revealed that DNA extraction method is a major driver of community differences and that its explanatory power (LLD: 10.48%, 500FG: 7.86%) was considerably higher than that of other anthropometric variables including sex (LLD: 0.34%, 500FG: 0.68%), age (LLD: 0.50%, 500FG: 0.67%) and BMI (LLD: 0.49%, 500FG: 0.35%). In contrast with earlier findings [9, 12, 22–25], paired samples isolated with different protocols showed lower similarity than unpaired samples extracted with the same protocol, as seen in the clustering on our PCoA. This discrepancy may be partially explained by the limited numbers of human participants included in the earlier comparative analyses (which ranged from one to 18 participants) compared to the 745 paired stool samples included in our analysis. These low numbers of participants substantially limited the ability to compare inter-subject and technical variation, thus hampering extrapolation of conclusions to large-scale analyses. Nevertheless, some studies have already reported results where the technical variability could be at least comparable to inter-individual variation at taxonomic [26] and functional level [9]. In addition, we found that the inter-individual distances had a limited sensitivity to the heterogeneity introduced by the DNA extraction method, as distance matrices of differentially extracted samples were positively correlated.
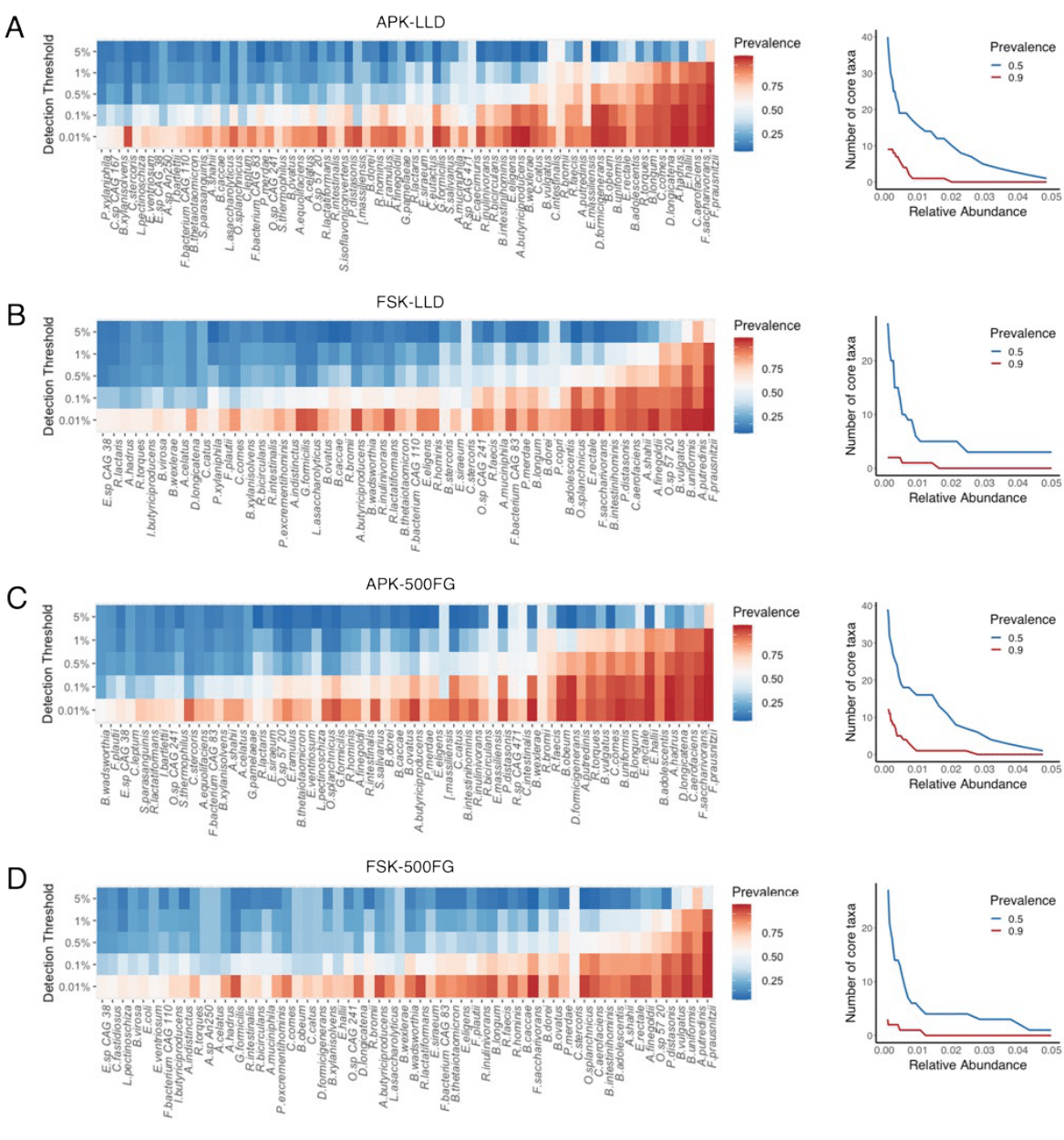
Several studies have previously described differences in taxonomic composition associated with DNA extraction protocol. However, they have mainly been restricted to genus-level taxonomic resolution due to the limitations of 16S rRNA gene amplicon–based analysis, the predominant method used in existing literature [8, 10, 12, 27, 28]. Evidence for substantial species-level heterogeneity in the human microbiome, and even for the presence of strain-specific phenotypes and functional profiles, highlights the need to gain deeper insights into lower taxonomic levels [29]. Therefore, in the present study, we report a massive alteration of species-level abundances in stool samples processed with APK and FSK, with a large proportion of the identified species being differentially abundant according to the DNA extraction protocol used. Interestingly, due to the compositional nature of microbiome data, protocol-dependent efficiencies in the disruption of the cell walls of gram-positive bacteria resulted in a considerable fraction of the differentially abundant species being gram-negative bacteria. Nonetheless, our findings broadly support previous work showing an increased abundance of gram-positive bacteria after the inclusion of a mechanical lysis step. Remarkably, we also found that methodological differences in DNA isolation impacted phenotypic associations with diversity measures. For instance, DNA extraction with the APK method led to a loss of significance in the associations between microbial composition and sex, cholesterol level (LLD cohort) and consumption of vegetables (500FG cohort). In contrast, LLD samples extracted with this protocol showed a significant association of several lifestyle and dietary habits with microbial diversity (smoking, alcohol, meat, vegetables and fruit consumption) and composition (glucose level, physical activity and fruit consumption) that were not observed in FSK samples. Additionally, the contrasting DNA extraction efficiency of both protocols resulted in notable alterations in significant phenotypic associations with microbial taxa abundances. When focusing on prevalent bacterial species, we found that the differential abundances of Prevotella copri and Alistipes putredinis in samples extracted with both protocols (both species were more abundant in FSK) led to changes in the ability to identify associations with human phenotypes. Indeed, we observed a significant association between P. copri and sex only in FSK samples, whereas A. putredinis was associated to the same phenotype only in APK.
This study has several limitations. Despite our efforts to limit technical variability that could complicate the assessment of differences directly caused by DNA extraction protocol, our samples are subject to technical heterogeneity at several levels. Firstly, while the isolation of LLD samples was done at the same time and place for both protocols, FSK samples’ isolation with the APK and FSK methods was done in different laboratories and four years apart. Although only a minor effect size of storage time of extracted DNA has been previously reported [30, 31], inter-laboratory differences have been shown to impact microbial profiles, while having a limited effect on relative diversity levels [13]. Due to the absence of technical replicates, we cannot evaluate the variation introduced by the cross-lab effect. Secondly, effectively assessing the impact of the bead-beating step would require a comparative analysis of both DNA extraction protocols with and without this additional step, so the design of the current study did not allow us to disentangle the effect of this step from that of the rest of the extraction procedure. Lastly, samples extracted with each protocol were sequenced in two different sequencing centers. While both centers applied the same sequencing technology, this difference could still represent another layer of technical variation, although previous studies have described sample sequencing to have a smaller impact than DNA extraction method [14].
Notwithstanding these limitations, our study expands upon previous findings that the DNA extraction procedure used has a large impact on the gut microbial diversity and structure recovered. To our knowledge, our analysis is the largest study to assess the impact of DNA extraction method and fecal sample processing on recovered gut microbiome profiles, and our considerable sample size overcomes the statistical power limitations of earlier comparative analyses. This, in combination with the use of shotgun metagenomics and the inclusion of the latest Metaphlan database release in our analysis, provides evidence of disruption of the species-level microbial profile by alternative DNA extraction methods. Although we only tested the differences in stool samples, it is likely that sufficient detection power will make it possible to unravel a similar effect in other microbiome samples, including those with lower biomass. In addition, we have demonstrated how the technical variability effect translates into the phenotype association analysis, pinpointing the influence of DNA extraction methodology on biological conclusions. This finding may help explain the considerable heterogeneity and low replication rate found in microbiome studies to date, an issue that has greatly hampered clinical translation of microbiome research findings.
{kind=link}
{kind=link}
{kind=link}
{kind=link}