Expression of 1C metabolism-associated genes
To evaluate the biological function of 1C metabolism-associated genes in the occurrence and development of LUAD, the expression pattern of 26 1C metabolism-associated genes was assessed in LUAD and adjacent normal tissues. Significant differences were observed in the expression levels of 23 genes between LUAD and adjacent normal tissues (Figure 1A). The expression level of 20 genes was upregulated, including PSAT1, PSPH, FTCD, SHMT1, SHMT2, MTHFD2L, MTHFD2, MTHFD1L, MTHFD1, GCAT, DMGDH, ALDH7A1, CHDH, TYMS, GART, ATIC, ALDH1L1, ALDH1L2, DHFR and MTFMT (Figure 1B). GNMT and MTHFR were significantly reduced in LUAD compared to adjacent normal tissues (Figure 1B). These results suggest that 1C metabolism-associated genes have important biological roles in LUAD development.
Prognostic value of 1C metabolism-associated genes
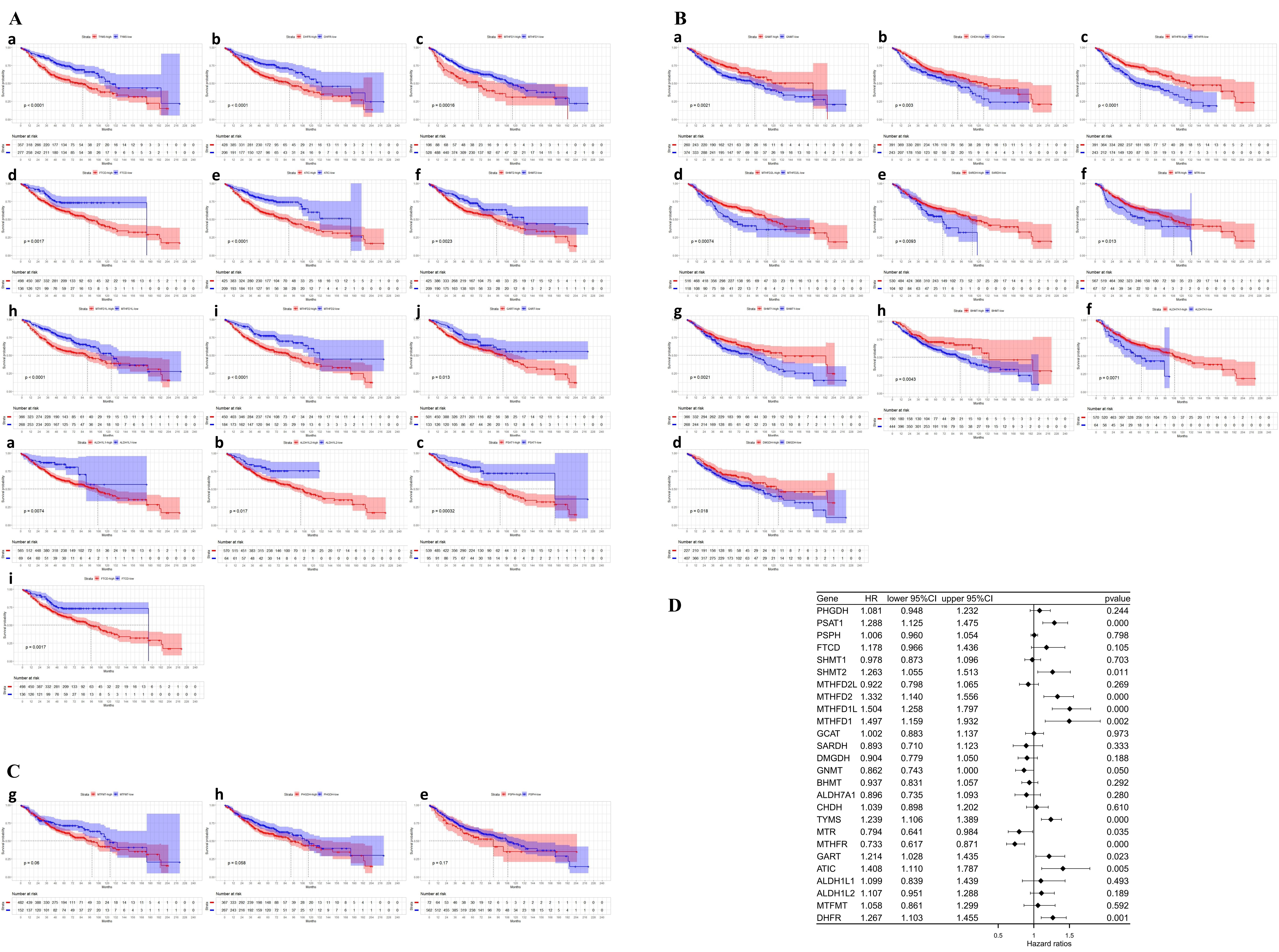
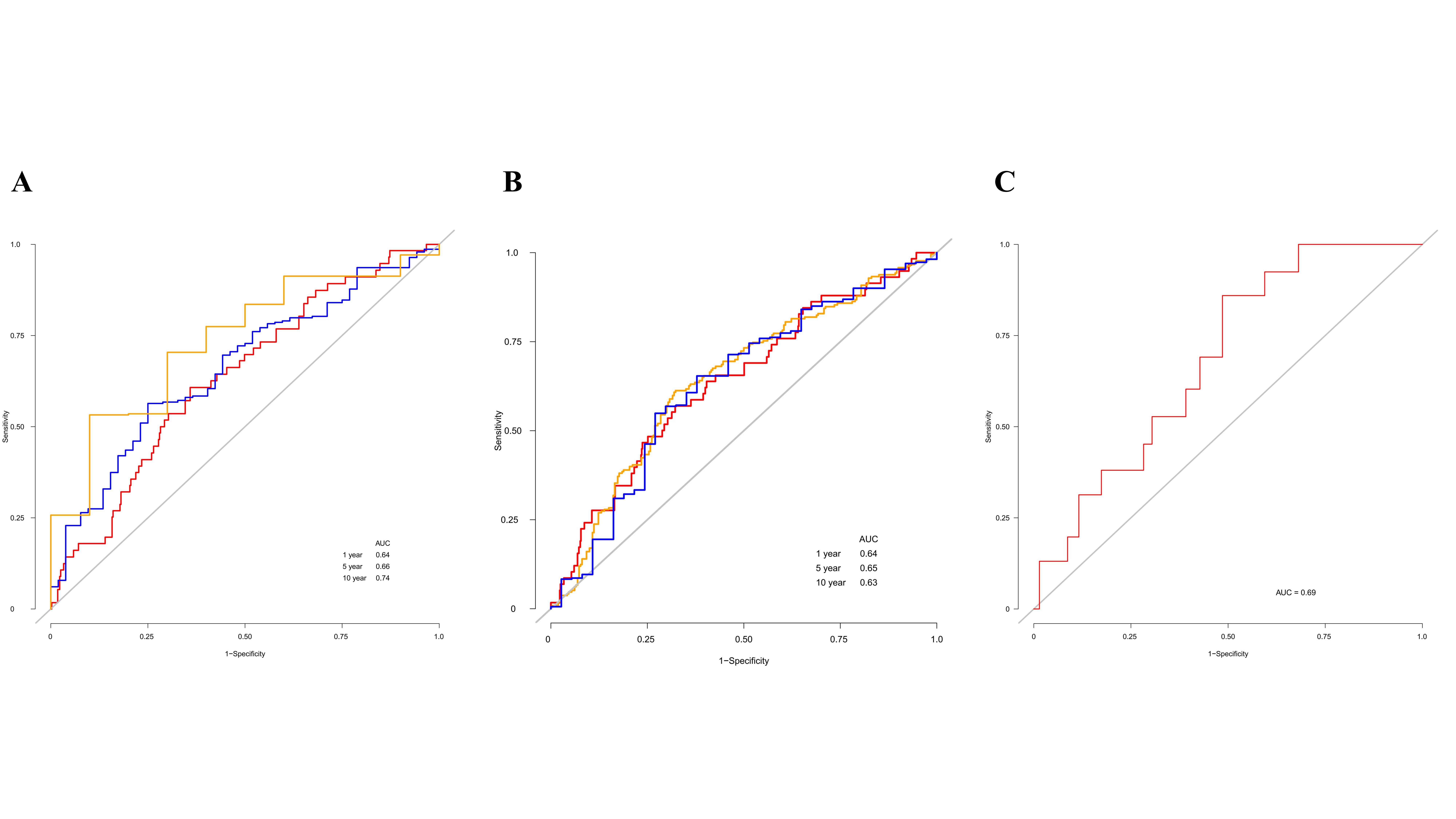
We further investigated the prognostic significance of 1C metabolism-associated genes in patients with LUAD in TCGA cohort. The Kaplan-Meier analysis based on an optimal cutoff shows that 17 genes were associated with OS, while nine genes were unrelated to prognosis (Figure 2A-C). The nine genes were identified as risk factors and included ATIC, GART, MTHFD1, MTHFD1L, MTHFD2, PSPH, SHMT2, DHFR, and TYMS (Figure 2A). Several other genes were considered protective factors, such as CHDH, GCAT, GNMT, MTHFD2L, MTHFR, MTR, SARDH, and SHMT1 (Figure 2B). A univariate Cox regression analysis was also performed and 10 genes had a significant prognostic correlation with OS. MTHFD2, MTHFD1L, MTHFD1, TYMS, DHFR, and ATIC were risk factors, while SARDH, CHDH, GNMT and MTHFR were protective factors (Figure 2D). In GEO cohort, Kaplan-Meier analysis based on the optimal cut-off revealed that 23 genes were related to OS, including 13 risk factors and 10 protective factors (Supplementary Figure 1A-C). Univariate Cox regression analysis was also performed and 11 genes were observed associated with OS (Supplementary Figure 1D). In addition, ROC curves were drawn to assess the specificity and sensitivity of 1C metabolism related-genes. The results indicated that the value at 1-, 5- and 10-year were 0.63, 0.67 and 0.74 in TCGA cohort, respectively (Supplementary Figure 4A). The AUC value of 1-year, 5-year and 10-year in GEO cohort were 0.64, 0.65 and 0.63, respectively (Supplementary Figure 4B).
1C metabolism-associated gene-based consensus clustering
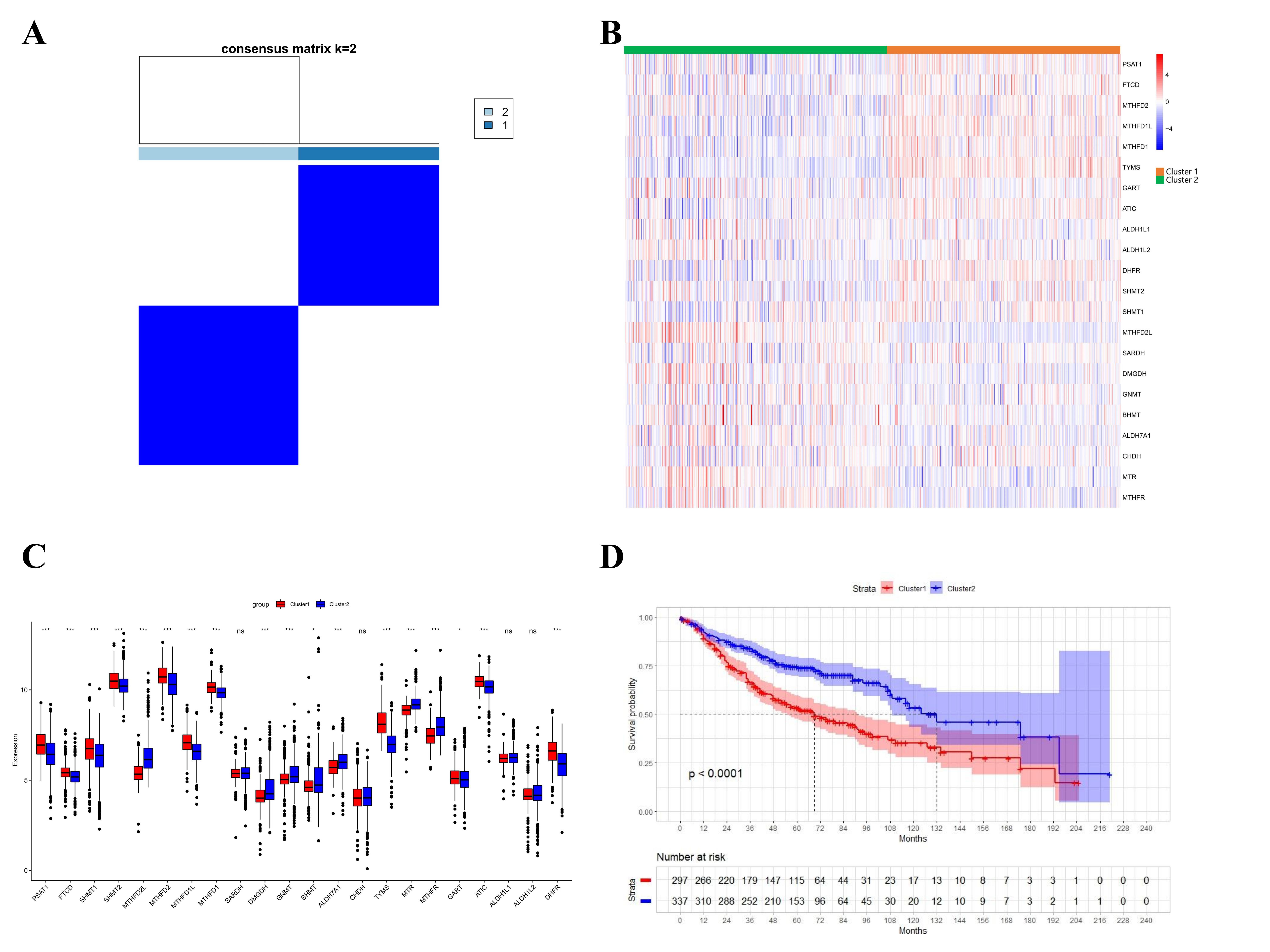
Consensus clustering was performed to investigate 1C metabolism-associated gene heterogeneity in TCGA cohort. A total of 497 patients with LUAD were clustered into two subtypes. Cluster 1 (n=248) was characterized by a high expression of high-risk genes while cluster 2 (n=249) was identified by a high expression level of protective genes (Figure 3A). These two clusters exhibited the opposite expression pattern. Cluster 1 was characterized by high expression of PHSH, SHMT2, MTHFD2, MTHFD1L, MTHFD1, TYMS, GART, ATIC, and DHFR, as well as low expression of SHMT1, SARDH, GNMT, CHDH, MTR and MTHFR (Figure 3B, C). A Kaplan-Meier analysis showed that patients who were divided into the cluster 1 subgroup suffered inferior OS (median OS: 41 vs. 60 months, p=0.0003; Figure 3D). Clinical characters between the two clusters were also investigated. Tumor metastasis (p=0.016), advanced stage (p=0.036), and smoking status (p<0.001) were more frequently observed in cluster 1 (Supplementary Table 1). In addition, consensus clustering was also performed in GEO dataset, and two clusters were identified, including cluster 1 (n = 397) and cluster 2 (n = 437) (Supplementary Figure 2A). Compared with TCGA cohort, similar expression patterns in two clusters were observed in GEO dataset (Supplementary Figure 2B, C). Kaplan-Meier analysis also revealed that cluster 1 subgroup suffered inferior OS in GEO cohort (median OS: 69 vs. 132 months, p<0.0001; Supplementary Figure 3D).
Consensus clustering-based genetic landscape and GSEA
To further investigate the genetic landscape differences between the two subtypes, somatic mutation data in LUAD patients were used. In cluster 1, TP53 was the most commonly mutated gene—with a frequency of 63%—followed by TTN, CSMD3, MUC16, and RYR2 (Figure 4A). In cluster 2, the top five mutated genes with a relatively low mutation rate were TTN, TP53, MUC16, KRAS, and RYR2 (Figure 4B). Although TP53 was one of the most frequently mutated genes in both groups, the mutation rate was significantly different between cluster 1 and cluster 2 (63% vs. 33%; Figure 4C). In addition, different mutation frequencies of the same gene between the two clusters were also observed for TTN, CSMD3, LRP1B, ZFHX4, and XIRP2 (Figure 4C), and the tumor mutation burden (TMB) of cluster 1 was significantly higher than in cluster 2 (Figure 4D).
GSEA analysis was used to investigate the transcriptomic alterations between these two groups. The most prominent gene ontology terms in cluster 1 were cell cycle, cell cycle procession, chromosome segregation, mitotic cell cycle, and nuclear chromosome segregation (Figure 4E).
Consensus clustering-based immune infiltrate analysis
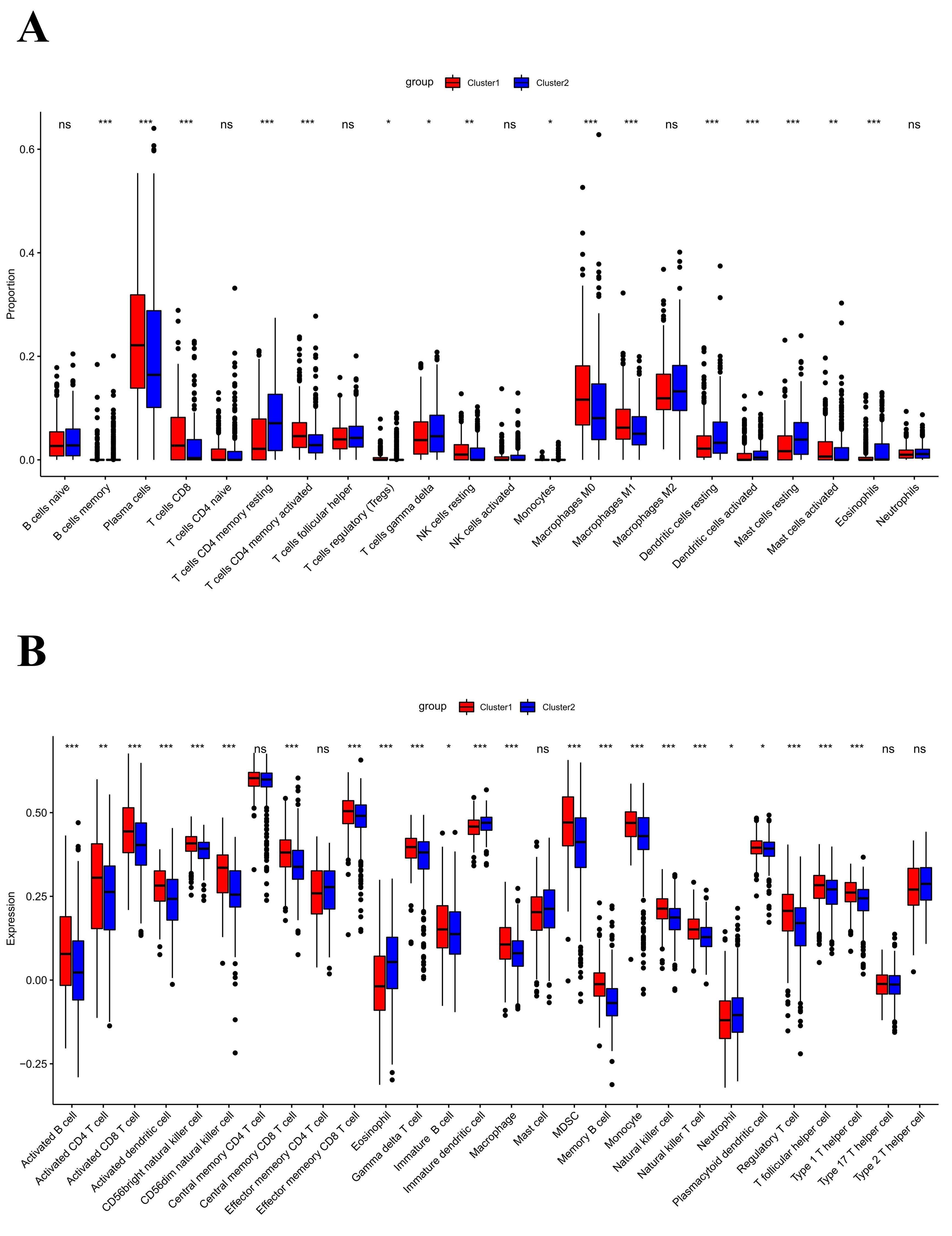
The infiltration level of immune cells in the TME has been confirmed to play an important role in tumor progression and immunotherapy. To evaluate the difference in immune cell infiltration between the two subgroups, CIBERSORT and ssGSEA were performed in TCGA cohort. The CIBERSORT analysis showed that CD8+ T cells, activated CD4 T cells, M0 macrophages, and M1 macrophages were significantly upregulated in cluster 1, while memory B cells, CD4 memory resting T cells, regulatory T cells, and monocytes were downregulated (Figure 5A). The ssGSEA analysis revealed that activated CD4 T cells, activated CD8 T cells, NK cells, effector memory CD4 T cells, memory B cells, natural killer T cells, and Type 2 T helper cells were significantly upregulated, and Type 17 T helper cells were significantly downregulated, in cluster 1 (Figure 5B). Moreover, the expression of PD-1 and PD-L1 was also upregulated in cluster 1 (Figure 5C, D). CIBERSORT and ssGSEA were also performed in GEO cohort. It was found that the infiltration level of immune cells in cluster 1 was higher than in cluster 2 (Supplementary Figure 3A, B).
Correlation analysis of methylation enzymes with 1C metabolism-associated genes
1C metabolism supports the biosynthesis and methylation of DNA and RNA by transferring 1C units. To explore the involvement of methylation with 1C metabolism-associated genes, 49 methylation enzymes were selected from previous studies32-35. In addition, we further evaluated the correlation of methylation enzymes with 1C metabolism-associated genes. The results revealed that the expression of methylation enzymes was significantly positively associated with 1C metabolism-associated genes, such as TYMS, MTR, MTHFR, SHMT2, MTHFD2L, MTHFD2, MTHFD1L, MTHFD1, GART, ATIC, PSAT1, PSPH, DHFR, and FTCD (Figure 6A). We also investigated the difference in DNA methylation levels between the two groups and observed a significant downregulation of DNA methylation in cluster 1 (Figure 6B). In addition, a further differential analysis revealed that hypermethylation of SEPT9 and KLF13 was foundin cluster 1, and hypomethylation of HNRNPR was also observed (Figure 6C).
1C metabolism-associated gene-based drug sensitivity analysis
To investigate the potential correlation between 1C metabolism-associated genes and drug sensitivity in multiple human tumor cell lines, a correlation analysis was performed in the CellMiner™ database. Cells with the expression pattern of cluster 1 were negatively associated with drug sensitivity to gemcitabine, oxaliplatin, obatoclax, imiquimod, and vorinostat (Figure 7A (a-e)), and positively correlated to drug sensitivity to 6-mercaptopurine, vandetanib, copanlisib, AT-9283 and byproducts of CUDC-305 (Figure 7A (f-g)). Cells with the expression pattern of cluster 2 were negatively correlated with drug sensitivity to etoposide, lapatinib, tepotinib, 6-thioguanine, and uracil mustard (Figure 7B (a-e)), but were positively correlated with drug sensitivity to paclitaxel, carboplatin, okadaic acid, pazopanib and alisertib (Figure 7A (a-e)). Patients in cluster 1 were also insensitive to paclitaxel and carboplatin, suggesting that patients in cluster 1 are likely resistant to gemcitabine, paclitaxel, oxaliplatin, and carboplatin treatment.
1C metabolism-associated genes are positively correlated with immunotherapy sensitivity
According to the results above, cluster 1 in the LUAD cohort is resistant to chemotherapy but may be sensitive to immunotherapy. We therefore explored the relationship between 1C metabolism-associated genes and immunotherapy in the IMvigor210 cohort. Consensus clustering was also performed, and two clusters (cluster 1 and cluster 2) were identified among patients in the IMvigor210 cohort (Figure 8A). A Kaplan-Meier analysis showed that for patients treated with immunotherapy, cluster 1 had a superior OS compared with cluster 2 (median OS: 11.2 vs. 7.8 months, p=0.0034; Figure 8B). The expression pattern of cluster 1 in the IMvigor210 cohort was similar to that of cluster 1 in the LUAD cohort (Figure 8C, D). A Kaplan-Meier analysis revealed that high expression of DHFR, TYMS, GART, MTHFD2 and SHMT1 were correlated with a superior OS (Figure 8E). In addition, the expression levels of TYMS, GART, and MTHFD2 in patients with a complete or partial response were higher than for patients with stable or progressive disease (Figure 8F). In addition, ROC curves were drawn to assess the specificity and sensitivity of 1C metabolism related-genes with the value of 0.69 (Supplementary Figure 4C).
{kind=link}
{kind=link}
{kind=link}
{kind=link}