PRDX4 specifically locates on macrophages of silicosis patients and upregulates the secretion of inflammatory factors.
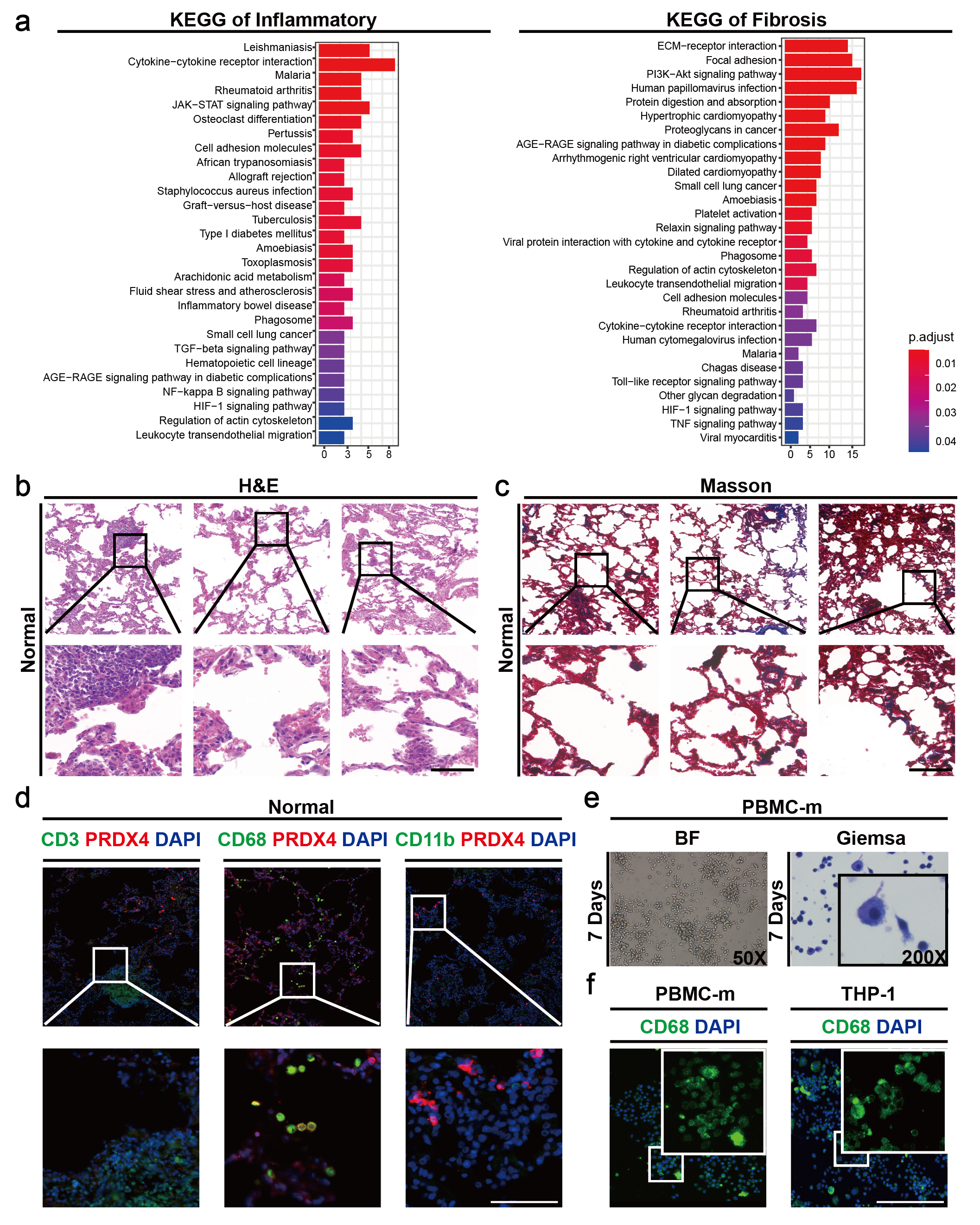
To determine the cellular localization and function of PRDX4 in silicosis patients, we downloaded RNA-seq data of lung tissue from silicosis patients19, and screened 201 inflammation and 307 fibrosis genes related to silicosis progression from the GSEA database (Supplementary Table 1). It was revealed that 36 inflammatory genes and 95 fibrosis genes were differentially expressed in normal and silicosis lung tissues. Furthermore, most inflammatory genes such as IL1, IL10, IL11, etc. and fibrosis genes like COL1A1, COL3A1, etc. were highly expressed in silicosis tissue (Supplementary Table 2). However, in PRDXs family, only PRDX4 was differentially highly expressed in silicosis tissue, which was positively correlated with the inflammatory and fibrotic factors (Fig. 1.a). In addition, KEGG analysis demonstrated that inflammatory genes were mainly enriched in JAK-STAT, cell surface receptor and TGF receptor signaling pathways, whereas fibrosis genes were mainly enriched in PIFK-AKT, ECM receptor and AGE-RAGE pathways, suggesting that these pathways play an important role in regulating the process of silicosis (Supplementary Fig. 1.a, Supplementary Table 3).
To further investigate the pathological changes in silicosis lung tissue, three pairs of clinical specimens from silicosis patients were detected. Hematoxylin and eosin (H&E) staining showed that lots of nodules and inflammatory cells were generated and aggregated in silicosis tissue and silicon particles were mainly aggregated around AMs (Fig. 1.c), while no significant pathological changes were observed in normal lung tissues (Supplementary Fig. 1.b). In addition, extensive fibrotic and collagenous substances were produced in silicosis tissue detected by Masson staining (Fig. 1.d), but no significant changes in normal lung tissue (Supplementary Fig. 1.c). To identify the cellular localization of PRDX4, we detected human normal and silicosis tissues by immunofluorescence, and found a large amount of AM, T cell and neutrophil infiltration in silicosis tissues. But PRDX4 was only highly expressed in AMs (Fig. 1.b, Supplementary Fig. 1.d). The results indicated that the progression of silicosis would be accompanied by the increase of immune cells like AMs, especially, and the high expression of PRDX4 was a key character of AMs. To illustrate how PRDX4 affect the AMs of silicosis patients, we selected human myeloid leukemia monocyte THP-1 and human peripheral blood mononuclear cell PBMC-m, and induced them to differentiate into macrophages respectively. PBMC-m was induced by M-CSF (100 ng/mL) for 7 days and identified by Giemsa staining and CD68 immunofluorescence labeling (Supplementary Fig. 1.e-f). THP-1 was induced by PMA (180 ng/mL) and identified by CD68 immunofluorescence labeling (Supplementary Fig. 1.f). Then, the two cells were simulated by CS (50 mg/cm2) for 6 h, 12 h and 24 h. It was found that both cells gradually changed from smooth round to shrunken oval morphologically, and the CS around gradually decreased (Fig. 1.e-f). Further RT-qPCR detection showed that PRDX4 and inflammatory factor-related genes such as TNF-α, TGF-β, IL1α, IL1β and IL6 all up-regulated in a time-dependent manner, and their expression was positively correlated (Fig. 1.e-f). Therefore, it can be concluded that PRDX4 specifically localized on AMs and promoted the secretion of inflammatory-related factors in silicosis patients. While, the function of PRDX4 in AMs still needs to be further clarified.
PRDX4 is positively correlated with the process of inflammation and fibrosis in silicosis mice.
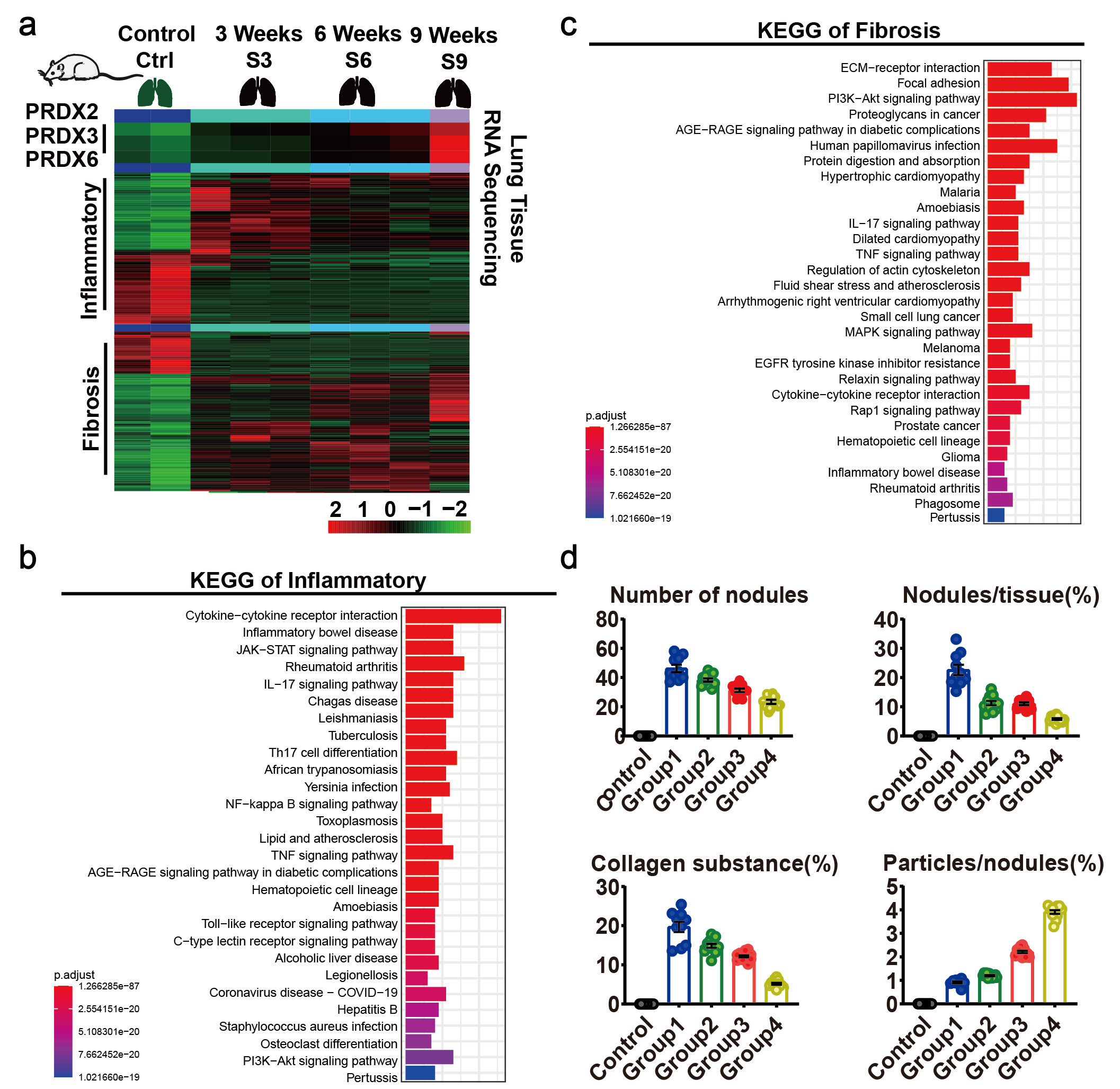
We downloaded RNA-seq data of lung tissue from silicosis mice in different stages17, and screened 201 inflammation and 307 fibrosis genes related to silicosis progression from the GSEA database (Supplementary Table 1). It was revealed that 109 inflammatory genes and 195 fibrosis genes were differentially expressed in lung tissues between normal and silicosis mice (Supplementary Table 4). With the progression of silicosis, the expression of inflammatory genes gradually decreased, while the fibrosis genes increased (Supplementary Fig. 2.a). It was revealed by KEGG pathway enrichment analysis that inflammatory genes were mainly enriched in inflammatory factor regulatory and JAK-STAT signaling pathways, and fibrosis genes were primarily enriched in ECM, PI3K-AKT and AGE-RAGE pathways, suggesting that these key pathways might affect the occurrence and development of silicosis (Supplementary Fig. 2.b-c, Supplementary Table 5). The dataset we used only contains PRDX2, RPDX3 and PRDX6 in the PRDXs family, which were overexpressed in the lung tissue of silicosis mice, and their expression gradually increased with the aggravation of silicosis (Supplementary Fig. 2.a). However, the correlation between other members of PRDXs (PRDX1, PRDX4 and PRDX5) and the process of silicosis remains unclear.
In order to dynamically observe the correlation between PRDXs and the expression of inflammation and fibrosis-related genes, the silicosis mouse model was constructed. we instilled CS in the nasal cavity of mice with normal saline as a control, and the mice were sacrificed after 1, 7, 14, 21 days that were correspondingly classified as groups 1 to 4 (Fig. 2.a). Compared with the control group, the weight of mice in the CS groups dropped significantly, but when stopping instilling CS, their body weight gradually increased (Fig. 2.b). The lung volume of control group was the smallest and the surface was smooth, the lung of group 1 seemed the largest with gritty protrusions, however, the lungs in other groups remained unchanged (Fig. 2.c). These findings suggest that mice have self-healing function to alleviate the toxic effect when the stimulus of CS is withdrawn. H&E staining showed that there were no obvious pathological changes in the lung tissue of control group, while in group 1, a lot of nodules were generated, accompanied by large amounts of inflammatory cells aggregation (Fig. 2.c). Among these five groups, group 1 had the largest number and relative area of silicosis nodules, which were gradually decreased in group 2 to 4 (Supplementary Fig. 2.d). Consistently, the collagen in lung tissue gradually decreased with the prolongation of repair time (Supplementary Fig. 2.d). We also found that silica particles (indicated by the white arrow) were mainly aggregated in the silicosis nodules using polarization analyses, and the quantity of silica particles was increased with repair time, which may be a self-healing method of silicosis mice (Fig. 2.c). Together, we conclude that mice can repair themselves in a time-dependent manner after removing the stimulation of CS.
To study the relevance of PRDXs to inflammation and fibrosis process in silicosis mice, we detected the expression of related genes in the lung tissues of mice. Compared with control group, inflammation and fibrosis-related factors were generally increased in the lungs of silicosis mice. However, inflammation-related genes such as TNF-α, TGF-β, IL6, IL1α and IL1β were increased firstly and then decreased with increasing repair time, and the peak appeared on the seventh day after cessation of CS stimulation (Fig. 2.d). While, the expression of fibrosis-related genes such as α-SMA, COL1A1 and COL3A1 were generally up-regulated, among which, α-SMA and COL3A1 peaked after 14 days (Fig. 2.d). As for PRDXs family, PRDX1, PRDX2, and PRDX4 all increased firstly and then decreased, wheras PRDX3, PRDX5, and PRDX6 did not change significantly (Fig. 2.e). Further pearson correlation analysis revealed that only PRDX1 and PRDX4 were highly correlated with inflammatory and fibrotic genes (Fig. 2.f), suggesting that PRDX1 and PRDX4 may play an important role in regulating the process of inflammation and fibrosis in silicosis mice.
PRDX4 specifically locates on macrophages of silicosis mice and upregulates the secretion of inflammatory factors.
To investigate the cellular localization of PRDX1 and PRDX4 in lung tissues of silicosis mice, we detected their expression in T cells (CD3+), macrophages (F4/80+) and neutrophils (Ly6g+) by immunofluorescence. It was found that PRDX4 was specifically highly expressed in AMs of silicosis mice (Fig. 3.a), while PRDX1 was not specifically localized (Fig. 3.b), indicating that PRDX4 plays a vital role in AMs. Therefore, we stimulated the mouse macrophage cell line (RAW 264.7) with CS (50 mg/cm2) for 6 h, 12 h and 24 h respectively, and found the cell morphology changed from smooth circles to shrunken ellipses and the SC around gradually decreased with the increase of stimulation time (Fig. 3.c), suggesting that macrophages can phagocytose CS particles. Meanwhile, the expression levels of PRDX4 and inflammatory factors TNF-α, TGF-β, IL1α, IL1β and IL6 detected by RT-qPCR were gradually up-regulated and positively correlated with the stimulation time of CS (Fig. 3.c). To gain insights into the effect of PRDX4 on the expression of inflammatory factors, first, we designed three PRDX4-specific knockdown siRNAs (si_Prdx4) and a negative control (si_NC) for mouse cells. Then, we selected one of the most effective si_Prdx4 (Supplementary Fig. 3.a) and verified that si_NC could not affect the expression of PRDX4 and related inflammatory factor genes in RAW 264.7 (Supplementary Fig. 3.b). Subsequently, we knocked down the PRDX4 in CS-induced RAW 264.7 cell and found that related inflammatory factors TNF-α, TGF-β, IL1α, IL1β and IL6 were significantly down-regulated (Fig. 3.d). These results indicate that the secretion of inflammatory cytokines in AMs of mouse induced by CS depends on the expression of PRDX4. While, the cellular localization and function of PRDX4 in silicosis patients deserves further investigation.
PRDX4 promotes AMs to secrete inflammatory-related factors through AKT/NF-κB pathway.
To study how PRDX4 regulates the secretion of inflammatory factors in AMs, the transcription factors (TFs) that control the expression of inflammation-related genes were searched in TRRUST database, and we found these TFs were enriched in NF-κB and JUN pathways (Fig. 4.a). In order to confirm whether NF-κB and JUN are involved in the expression of inflammation-related genes, we stimulated RAW 264.7, THP-1 and PBMC-m cells with CS (50 mg/cm2) for 24 h and found only the nuclear translocation of p65 was significantly increased (Fig. 4.b-c, Supplementary Fig. 4.a), suggesting that p65 may be a key TF regulating inflammation-related factors. To verify whether p65 is related to PRDX4, we first transfected RAW 264.7 with si_NC (ACGUGACACGUU CGGAGAATT) and si_Prdx4 (AUUAAUCCA GGCCAAGUGGTT), and then stimulated the cells with CS for 24 h. We found the nuclear translocation of p65 was significantly reduced when Prdx4 was knocked down (Supplementary Fig. 4.b), suggesting that PRDX4 regulates the activation of p65 in AMs. Consistently, specific p65 inhibitor PG490 could markedly inhibited the expression of TNF-α, TGF-β, IL-1α, IL-1β and IL6 in CS-stimulated Raw 264.7 cells, but had no effect on PRDX4 (Supplementary Fig. 4.c), indicating that p65 is located downstream of PRDX4 to regulate the transcription of inflammatory factors. Given the species differences between mice and humans, we further treated THP-1 and PBMC-m cells with a PRDX4 inhibitor (Conoidin A) and achieved the similar results that the nuclear import of p65 was blocked but normal for c-jun (Fig. 4.d-e). Therefore, we speculated that PRDX4 promoted the expression of inflammatory factors in macrophages through inducing p65 into the nucleus. To verify our conjecture, we inhibited the activity of PRDX4 with Conoidin A in CS-stimulated THP-1 and PBMC-m cells, and found the expression of inflammation-related factors such as TNF-α, TGF-β, IL-1α, IL-1β and IL6 were significantly reduced (Fig. 4.f-g). Also, considering that Conoidin A can inhibit PRDX2, we transfected THP-1 and PBMC-m cells with si_NC (UUCUCCGAACGUGUCAC GUTT) and si_PRDX2 (572, GGGCCUCUUU AUCAUCGAUTT) that was selected from three si_ PRDX2s (90, 379 and 572), and then the cells was stimulated with CS (Supplementary Fig. 4.d-e). Most inflammation-related factors expressed in macrophage with no changes when PRDX2 was knocked down (Supplementary Fig. 4.f-g). Taking the above data together, we found that PRDX4 could activate NF-κB and promote CS-simulated AMs to secrete inflammatory-related factors.
However, PRDX4 is not a kinase and cannot directly induce the phosphorylation and nuclear translocation of p65. We guessed that PRDX4 might depend on other kinases such as AKT and TAK1 to regulate p6520. To prove our speculation, we inhibited PRDX4 with si_PRDX4 or Conoidin A in CS-induced THP-1 and PBMC-m cells, respectively. It was found that p-AKT and p-p65 were significantly downregulated in PBMC-M cells, while p-AKT and p-TAK1 were both down-regulated in THP-1 cells (Fig. 4.h). To further clarify whether the activation of p65 is dependent on p-AKT, we treated cells with a selective AKT inhibitor MK-2206 and found that both p-AKT and p-p65 were still significantly downregulated (Fig. 4.i), indicating that PRDX4 can regulate the secretion of inflammatory-related factors in CS-induced AMs through AKT/NF-κB pathway.
CS-simulated AMs promote the differentiation of epithelial cells into myofibroblasts in vitro.
The above data confirms that PRDX4 plays a critical role in activating AMs of silicosis mice and patients through AKT/NF-κB pathway, but the correlation of PRDX4 and fibrosis-related genes is still unclear (Fig. 1.a, Fig. 2.f). As we all known, the immediate cause of silicosis fibrosis is the deposition of excessive extracellular matrix (ECM), which is mainly generated from myofibroblasts21. Myofibroblasts are transformed from fibroblasts and alveolar epithelial cells (AECs)22, the process known as FMT and EMT, respectively. Therefore, we speculated that CS-stimulated AMs could promote the transformation of fibroblasts and AECs into myofibroblasts and ultimately lead to extensive fibrosis in the lungs (Fig. 5.a). Thus, we designed macrophage/epithelial (RAW264.7/MLE12, THP-1/A549 and PBMC-m/A549) and macrophage/fibroblast (THP-1/WI-38, PBMC-m/WI-38) transformation models separately. We cultured mouse lung epithelial cell MLE12 and human lung epithelial cell A549 with conditioned medium for 0, 24 and 48 h, and the medium was collected from CS-stimulated mouse macrophage (RAW 264.7) or human macrophages (THP-1 and PBMC-m). It was found that the migration and proliferation abilities were significantly enhanced detected by scratch test and Giemsa staining assay, respectively, and the expression of COL1A1 and COL3A1 were significantly increased tested by RT-qPCR (Fig. 5.b-d). It could be concluded that the inflammatory factors secreted by CS-stimulated macrophages from mouse or human promote the transformation of epithelial cells into fibroblasts. To further clarify the role of activated macrophages in promoting FTM, we designed THP-1/WI-38 and PBMC-m/WI-38 cell transformation models. After culturing human-derived lung fibroblasts (WI-38) with conditioned medium from CS-induced THP-1 or PBMC-m cells for 0h, 24h and 48h, the migration and proliferation of WI-38 were significantly enhanced, and the expression levels of α-SMA, COL1A1 and COL3A1 were markedly up-regulated (Fig. 5.e-f), which further demonstrated that cytokines produced from CS-stimulated macrophages could induce FMT.
PRDX4 inhibitors reverse early silicosis and accelerate repair process.
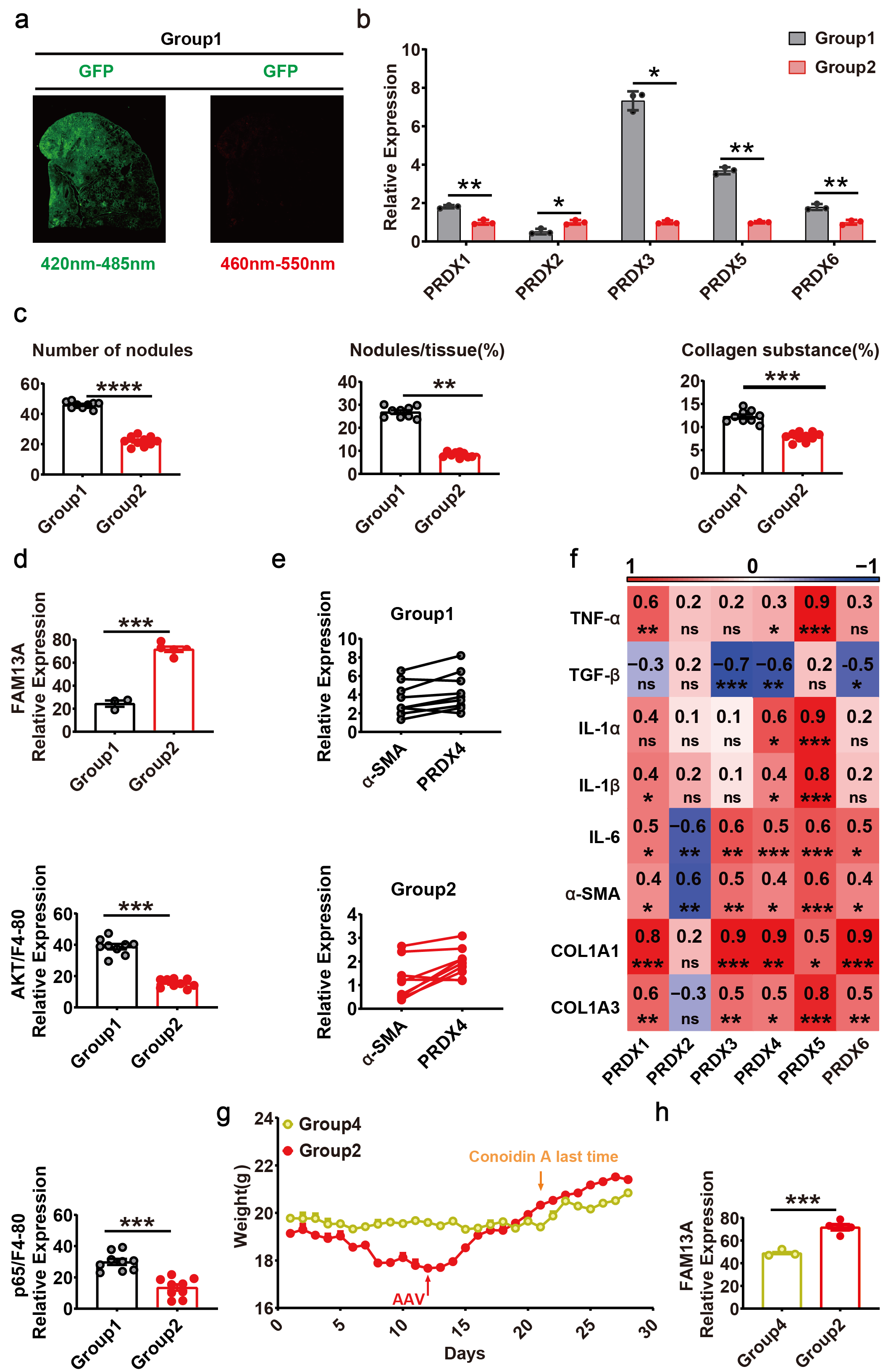
Given that PRDX4 induces AMs to secrete inflammatory factors through AKT/NF-κB pathway and promotes the progression of pulmonary fibrosis, PRDX4 may be a potential therapeutic target for pulmonary fibrosis. To determine whether blockade of PRDX4 could ameliorate the development of pulmonary fibrosis, we designed a mouse model of PRDX4 inhibitor (Conoidin A) to reverse early silicosis. The model consisted of 4 groups of mice (Fig. 6.a) including CS treatment for 21 days (Group1), CS + Conoidin A treatment for 21 days (Group2), CS treatment for 21 days followed by self-healing for 21 days (Group3), and CS + Conoidin A treatment for 21 days followed by self-healing for 21 days (Group4). It was obvious that Conoidin A markedly improved the weight of mice in group 2 and mitigated the toxicity of CS compared with control group 1 (Fig. 6.b). After stopping the stimulation of CS on day 21, the body weight of mice in group 3 also began to increase (Fig. 6.b), indicating that the mice had a certain self-recovery function. However, after 21 days, the weight recovery of mice that was administered with Conoidin A in group 4 was more obvious (Fig. 6.b), which further indicated that inhibition of PRDX4 in vivo could significantly accelerate the recovery of lung function in silicosis mice. Moreover, mice treated with Conoidin A showed smaller lungs with no gritty protrusions than that of group 1(Fig. 6.c). Compared with the control (group 1), mice in group 3 and 4 had fewer lung nodules and aggregated inflammatory cells, and mice in experimental groups (group 2, 3 and 4) showed a gradually decreasing amount of collagen substance, as revealed by H&E staining and Masson’s trichrome staining (Fig. 6.c), which was consistent with the statistical results of the pathological analysis (Supplementary Fig. 5.a). To further clarify the effect of PRDX4 inhibition on lung inflammation and fibrosis, we detected the expression of related genes. The inflammation-associated factors (TGF-β, IL1α, IL1β and IL6) and fibrosis-associated factors (COL1A1 and COL3A1) were suppressed in Conoidin A treatment groups, whether in the CS stimulus stage (Group 1 vs Group 2) or the repair stage (Group 3 vs Group 4). Besides, the expression of fibrotic markers α-SMA in the stimulus stage and TNF-α in the repair stage were also suppressed (Fig. 6.e). We then analyzed the expression of PRDX genes and found PRDX4 was significantly inhibited by Conoidin A during the repair stage (Fig. 6.d, Supplementary Fig. 5.b), suggesting that the expression of PRDX4 may be regulated by positive feedback. Overall, we proved that inhibiting PRDX4 could prevent lung inflammation and fibrosis progression in silicosis mice.
To evaluate the lung function of mice, we detected the expression of the lung function marker FAM13A24, and found that Conoidin A can significantly up-regulate the expression of FAM13A (Fig. 6.f), suggesting that blocking PRDX4 can accelerate the repair of lung function in silicosis mice, which was consistent with the results of pathological analysis (Supplementary Fig. 5.c). Subsequently, we analyzed the expression of AKT and p65 in AMs (F4/80) of the lung tissues, and it was shown that Conoidin A significantly inhibited the activation of AKT and p65, indicating that PRDX4 regulates AMs through the AKT/NF-κB pathway (Fig. 6.f, Supplementary Fig. 5.d-e). Meanwhile, plenty of AMs with PRDX4 high-expressed clustered around myofibroblasts (Fig. 6.f), and the expression of PRDX4 and α-SMA were positively correlated (Supplementary Fig. 5.f).
Specific knockdown of PRDX4 protein in AMs alleviates silicosis.
Although Conoidin A could inhibit PRDX4 and improve the self-repair of lung function in vivo, it was unable to prove that the PRDX4 inhibition was in AMs, as intraperitoneal injection is a systemic mode of administration. Thus, we verified that AAV viral vector could efficiently infect the lungs of mice with the method of nasal intubation drip (Supplementary Fig. 6.a). And we inserted the macrophage-specific promoter sp14624 into the vector and designed AAV_SP146_shPRDX4 whose PRDX4 was specifically knocked down (Fig. 7.a). Next, we stimulated mice with CS at day 0, 3, 6 and 9 sequentially and transfected with AAV_SP146_ shPRDX4 at day 12 (Fig. 7.b). Compared with the control group, the weight of mice in the transfected group started to increase at day 13 (Fig. 7.c), the lung was smaller and smoother, the nodules, inflammatory cells and collagen material were significantly reduced (Fig. 7.d), which were consistent with the results of pathological statistical analysis (Supplementary Fig. 6.c). These data suggested that stimulation with CS lead to lung inflammatory cell infiltration and promoted lung fibrosis, which could be alleviated by knocking down PRDX4 in AMs during the inflammatory stage. To confirm that PRDX4 was effectively knocked down by AAV_SP146_shPRDX4 in AMs, the macrophage marker (F4/80) and PRDX4 were labeled, indicating that the expression of PRDX4 was significantly downregulated (Fig. 7.e).
To further verify that pulmonary inflammation and fibrosis could be improved by conditional knockdown of PRDX4 in AMs, we detected the expression of related genes. It was shown that the expression of inflammation-related genes such as TNF-α, TGF-β, IL1α, IL1β and IL6 and fibrosis-related genes such as α-SMA, COL1A1 and COL3A were all markedly inhibited (Fig. 7.j). Besides PRDX4, the expression of other PRDXs including PRDX1, PRDX3, PRDX5, PRDX6 was also decreased when PRDX4 was conditionally knocked down in AMs (Fig. 7.g, Supplementary Fig. 6.b). However, only PRDX4 was highly correlated with inflammation and fibrosis genes as revealed by correlation analysis of gene expression, suggesting PRDX4 as a key gene that affects the process of inflammation and fibrosis in silicosis mice (Supplementary Fig. 6.f).
To clarify the recovery of lung function in silicosis mice with AAV_SP146_ shPRDX4, we detected the expression of FAM13A and found it was significantly increased in the lung tissues (Fig. 7.f, Supplementary Fig. 6.d). Furthermore, the expression of AKT and p65 in AMs (F4/80) of the two groups of mice was analyzed. It was shown that the activation of AKT and p65 in AMs were suppressed when PRDX4 was knocked down, indicating that PRDX4 regulates AMs through the AKT/NF-κB pathway (Fig. 7.f, Supplementary Fig. 6.d). Meanwhile, the expression of PRDX4 and α-SMA were positively correlated (Supplementary Fig. 6.e).
Remarkably, we found that conditional knockdown of PRDX4 (AAV_SP146_ shPRDX4) was superior to systemic inhibition of PRDX4 (Conoidin A) in silicosis mice, the weight gain and expression of FAM13A in the lungs of the former mice were significantly higher than those of the latter (Supplementary Fig. 6.g-h). In conclusion, PRDX4 was proved as a potent target for regulating the progression of lung inflammation and fibrosis, and targeted inhibiting PRDX4 in the early stage can effectively alleviates silicosis (Fig. 8).
{kind=link}
{kind=link}
{kind=link}