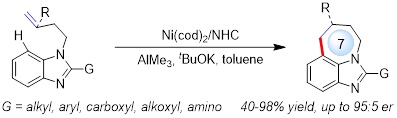
Reaction optimization. We commenced our study by selecting benzoimidazole 1a as a model substrate, nickel as a catalyst and Al-Lewis acid as a co-catalyst (Fig. 3). A systematic survey on Ni metals, Al Lewis acids, ligands, bases, and other reaction parameters led to the optimal conditions: 10 mol% of Ni(cod)2, 10 mol% of IPr·HCl, 10 mol% of AlMe3 and 40 mol% of tBuOK in toluene at 130 °C, under which an endo cyclization was exclusively achieved, providing tricyclic imidazole 2a bearing a 7-membered ring in 98% yield (Fig. 3, entry 1).
Control experiments showed that the combination of Ni, IPr, AlMe3 and tBuOK is critical, and the removal of any of them would greatly reduce the yield (entries 2−5). Traditional phosphine ligands such as monophosphines and bidentate phosphines were all ineffective (entries 6 and 7), whereas other N-heterocyclic carbenes were still compatible, albeit with a little lower yields (entries 8 and 9). In addition, in situ formed Ni(0) was also an effective catalyst, yet providing only 42% yield (entry 10). Base acted as another critical role in the reaction. tBuOLi was inefficient, whereas tBuONa worked well, affording a comparable result to that of tBuOK (entries 11 and 12). Notably, more than 10 mol% of tBuOK was essential to the reactivity (entries 13−15). The use of 10 mol% of tBuOK gave no products (entry 13), instead, leading to an imidazole with free NH group in 5% yield, which was formed from the decomposition of alkene-isomerization substrate. We reasoned that excess tBuOK could suppress the isomerization of the terminal alkene as the literature proposed.45
Scope of imidazoles and alkenes. With the optimized conditions in hand, various benzoimidazole motifs bearing different substituents on the aromatic ring were investigated first (Fig. 4). Results showed that either electron-donating groups such as methyl (2b) and tert-butyl (2c) or electron-withdrawing groups such as CF3O (2d), F (2e to 2h), CF3 (2i) and carboxylate (2j) were well compatible with the reaction, providing the corresponding products in 80−98% yield.
Notably, C2 substituents of benzoimidazoles proved critical to the reactivity. Without C2 substituents, C2−H cyclization would dominate to form a 6-memebered ring as we previously reported,41 further suggesting that C7−H bond was quite unreactive towards Ni catalysis. In general, electron-deficient CF3 group on C2 position can ensure high reactivity with using only 10 mol% of AlMe3 co-catalyst, while CF3 group was not indispensable, and it can be replaced by a broad range of other substituents such as alkyl (2k and 2l), (hetero)aryl (2m and 2n), carbamoyl (2o and 2p), alkoxy (2q) and amino (2r) groups, providing the corresponding products in 40−79% yield by tuning the amount of AlMe3. Pleasingly, imidazole-2-ones, which also widely exist in numerous bioactive compounds, were well compatible with the current reaction. When N-protecting groups varied from Me (2s), Bn (2t), PMP (2u) to Ph (2v), the corresponding products can be smoothly obtained in 64−90% yield. In consideration of two symmetrical N atoms in the molecule, dual C−H annulation was then investigated and a tetracyclic product (2w) bearing two 7-membered rings can be smoothly achieved, which is not easily accessed by traditional Friedel-Crafts reaction because the second acylation would be quite difficult. Besides simple aryl and alkyl groups, carboxylate group was also tolerated, providing a translocator protein inhibitor (2x) in 60% yield.1
Next, the compatibility of alkene motifs were investigated (Fig. 5). Although internal alkenes were ineffective because of big steric hindrance, various 1,1-disubstituted terminal alkenes proved to be effective. Different types of alkyls such as methyl (3a), linear n-butyl (3b), branched cyclohexyl (3c) and functionalized alkyl (3d) were well tolerated, delivering the corresponding products in 82−96% yield. Considering that the incorporation of aryl motifs can significantly increase the complexity of molecules, we examined various aryl substituted alkenes (3e to 3p). Results showed that these aryl alkenes bearing either electron-rich groups such as methyl (3f to 3h), tBu (3i), Ph (3j), methoxy (3k), and naphthyl (3l) or electron-deficient groups such as CF3O (3m) and F (3n to 3p) at different positions of the aryl ring all proceeded smoothly, providing the corresponding products in 50−90% yield.
Enantioselective attempts. For the synthesis of medium ring, a flexible large ring transition state would be involved, rendering the enantioselective control of such a reaction quite challenging.29−34 By surveying a wide range of chiral carbenes, we found that bulky AnIPE was the optimal ligand (see the Supporting Information for details).46−48 With this ligand, a series of substrates with various alkene motifs were then tested (Fig. 6). In general, various aryl groups were well compatible with the current reaction, providing the corresponding products in good yields and with 91.5:8.5 to 95:5 er (3e to 3q). However, alkyl groups, albeit still with good yields, would result in slightly decreased ee (3a, 3d, 3r and 3s) owing to bigger structural flexibility. The absolute configuration of major enantiomer of the product was assigned to be (R) by single crystal X-ray diffraction.
Reaction utility and mechanistic discussion. To demonstrate the utility of the current method, a gram-scale reaction of 1a was conducted, and a comparable yield was obtained under the standard conditions (Fig. 7a). In addition, tricyclic imidazole derivative 2s can be easily oxidized at the benzylic position to produce an intermediate 4 in 62% yield, which can be further transformed into various bioactive molecules such as β-2-adrenergic agonists and zilpaterol (Fig. 7b).11−14 To gain more insights into the reaction, relevant mechanistic experiments were conducted. Deuterium-labeling experiment showed that C7-D on the aromatic ring was completely transferred to the 7-membered ring, and moreover, no deuterium scrambling was observed at other positions (Fig. 7c), which suggested that an endo-insertion of alkene to Ni−H bond could proceed via an irreversible step. Both competitive experiment between equivalent moles of 1a and d4-1a and parallel reactions revealed significant kinetic isotope effect (kH/kD = 5.75, 4.81, respectively), indicating that the C−H cleavage could be the rate-determining step (Fig. 7d), and it could proceed via oxidative addition mechanism because direct H transfer pathway in general gives low kinetic isotope effect.35 In addition, 19F NMR spectra of stoichiometric reactions suggested that nickel could rapidly coordinate to the alkene motif of substrate 1a, and then initiate next C−H cleavage and alkene insertion (see the Supporting Information). On the basis of these facts, a plausible mechanism was proposed as below (Fig. 7e): substrate 1a coordinates with AlMe3 and nickel first, and then facilitates Ni-catalyzed C7−H bond cleavage via oxidative addition process. Subsequent irreversible endo-type alkene migratory insertion and reductive elimination generates the Al-coordinated product, which exchanges with another substrate 1a to initiate a next cycle.
{kind=link}