Understanding the noncovalent (weak) interactions between asphaltene molecules is the key to further comprehending the viscosity and aggregation behavior of asphaltenes. In the past, intermolecular interactions were characterized indirectly by calculating the radial distribution function and the numerical distribution of distances/angles between atoms, which are far less intuitive than the average reduced density gradient (aRDG) method. This study selected three representative asphaltene molecules (AsphalteneO, AsphalteneT, and AsphalteneY) to investigate the relationship between viscosity and weak intermolecular interactions. Firstly, a non-equilibrium molecular dynamics (NEMD) simulation was employed to calculate the shear viscosities of these molecules and analyze their aggregation behaviors. In addition, the types of weak intermolecular interactions of asphaltene were visualized by the aRDG method. Finally, the stability of the weak intermolecular interactions was analyzed by the thermal fluctuation index (TFI). The results indicate that AsphalteneY has the highest viscosity. The aggregation behavior of AsphalteneO is mainly face-to-face stacking, while AsphalteneT and AsphalteneY associate mainly via offset stacking and T-shaped stacking. According to the aRDG analysis, the weak interactions between AshalteneT molecules are similar to those between AshalteneO molecules, mainly due to van der Waals interactions and steric hindrance effects. At the same time, there is a strong attraction between AsphalteneY molecules. Additionally, the results of the TFI analysis show that the weak intermolecular interactions of the three types of asphaltene molecules are relatively stable and not significantly affected by thermal motion. Our results provide a new method for better understanding asphaltene molecules' viscosity and aggregation behavior.
Research Article
aRDG analysis of asphaltene molecular viscosity and aggregation behaviors based on NEMD simulation
https://doi.org/10.21203/rs.3.rs-1989230/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 07 Dec, 2022
Version 1
posted
You are reading this latest preprint version
Asphaltene
Shear Viscosity
NEMD
Aggregation
Weak interaction
aRDG
Asphaltene, the heaviest, densest, and most polar component in petroleum, is classified as a complex mixture of heavy organic molecules that are insoluble in n-alkanes, including n-heptane and n-pentane soluble in aromatic solvents, such as toluene and benzene1–3. The viscosity of heavy oil is closely related to its asphaltene content, and the high concentration of asphaltene is the main reason for the high viscosity of heavy oil4, 5. In addition, the aggregation behavior of asphaltene molecules is responsible for the deposition, emulsification, and high viscosity of heavy oil, which significantly affect its utilization and value.
Asphaltenes are complex mixtures of polycyclic aromatic hydrocarbons substituted with alkyl side chains and heteroatoms, such as nitrogen, sulfur, and oxygen, which are typical substituents in conjugated cores6, 7 In general, asphaltene molecules have a polycyclic aromatic core that is composed of approximately 4–10 aromatic rings and fatty side chains with a length of 3–7 carbon atoms8, 9. In the past decade, various research groups have proposed and studied different asphaltene molecular models to study the mechanism behind asphaltene properties. Sjöblom10 reviewed the different types of proposed asphaltene models and summarized the composition and properties of asphaltenes and the research methods and results for different types of model asphaltene compounds.
During the past few decades, people have been exploring the mechanism of asphaltene aggregation. In 1990, Hunter and Saunders11 summarized three rules for porphyrins: (1) π-π repulsion dominates the face-to-face geometry; (2) π-σ attraction dominates the side geometry; and (3) σ-σ attraction dominates the offset π-stacking geometry. Three stacking modes are shown in Fig. 1. Pacheco12 employed classical molecular dynamics (MD) to simulate asphaltene aggregation under vacuum at different temperatures and concluded that the aggregation behavior of asphaltene molecular dimers could also follow these rules.
Takanohashi13 used MD simulation to study the stability of asphaltene aggregates of three model molecules at high temperatures and observed that aliphatic side chains and heteroatomic functional groups contribute to the stability of trimers. Rogel14 used the average structure model to study the interaction force of the binding process between asphaltene and resin. The results indicate that the stabilization energy obtained by asphaltene and resin is due mainly to the van der Waals forces between molecules. In contrast, the contribution of hydrogen bonds is low. In addition, some researchers have studied the relationship between asphaltene molecular structure and aggregation behavior 1, 15, 16.
Because of its many sources, asphaltene has a complex and diverse molecular structure, which has caused controversy for many years4, 17. Schuler18 utilized atomic force microscopy (AFM) and scanning tunneling microscopy (STM) to perform molecular orbital imaging for more than 100 asphaltene molecules and proposed that the molecular structure of asphaltene is an aromatic core, substituted with heteroatoms and a variable number of alkyl side chains. Sedghi1 employed MD simulations to investigate the relationship between the aggregation and molecular structure of asphaltene. The results indicate that the interaction between aromatic nuclei of asphaltene molecules is the main driving force of asphaltene aggregation. Furthermore, Jian16 carried out a series of MD simulations to study the effect of aliphatic side chain length on the aggregation behavior of asphaltenes. The degree of aggregation is not monotonically related to the side chain length. Asphaltene molecules with short or long side chains can form dense aggregates, while those with medium-length side chains cannot. Ekramipooy19 employed density functional theory and MD simulations to study the effect of heteroatoms on the aggregation of model asphaltenes. The results show that heteroatoms in the fatty side-chain more effectively increase the aggregation. The heteroatoms in the middle of the fat side chain strengthen the CH…C dispersion interaction through carbon polarization.
Although the studies on asphaltene viscosity and asphaltene aggregation behavior alone have been numerous and intensive, there are few studies on the relationship between the two, which is an important research direction needed to understand the microscopic mechanism of asphaltene molecules with different viscosities, so this is one of the focuses of this study. In addition, most of these studies14, 19 perform indirect characterization by calculating radial distribution functions and numerical distribution of interatomic distances/angles to study the asphaltene interactions. Yang20 proposed a visual method to study weak interactions as a direct characterization tool, referred to as the reduced density gradient (RDG) or noncovalent interaction (NCI) method. The so-called weak interaction refers to various forms of interaction whose strength is weaker than covalent chemical bonds, such as van der Waals interactions, π-π stacking, hydrogen bonds, halogen bonds, and dihydrogen bonds. To further identify the characteristics of the interactions between asphaltene molecules in a dimer, Wang2 carried out NCI visualization analysis, focusing on the intermolecular interactions in the asphaltene dimer, and screened the intramolecular interaction of asphaltene, concluding that π-π interactions are the main driving force of asphaltene aggregation. Ekramipooya19 adopted the RDG method to study the effect of heteroatoms on self-aggregation at different positions in a model of the asphaltene structure. By comparing the relationship between the RDG and sign (λ 2) ρ of different asphaltene dimer models, Ekramipooya concluded that the heteroatoms at the X3 position (in the middle of the fat side chain) strengthened the CH…C dispersion interaction through carbon polarization. Yang21 proposed the averaged RDG (aRDG) method (also known as the aNCI method), which is an extension of the original RDG method that can be used to analyze multi-frame structures, especially in combination with MD simulation techniques, to study weak interactions in equilibrium dynamic environments. Wu22 investigated the aggregation behavior of three reactive dyes in water by MD simulations. The dye-water molecular interactions were analyzed by the RDG and aRDG methods, and the position, intensity, and type of the interactions were visualized. The results show that the main interactions between the dye and anions are van der Waals and π-π stacking interactions.
The aRDG method can be employed to study average weak interactions between small molecules in kinetic processes and receptor-protein, small peptide-small peptide, and molecule-solid surface cases23–26. At present, although MD simulations have been widely employed to investigate the molecular structure and aggregation behavior of asphaltenes, few studies have used the RDG method, especially the aRDG method, to examine the weak interactions between asphaltene molecules. Schuler18 obtained the fundamental structures of some asphaltene molecules by using AFM. In this study, three representative structures of asphaltene molecules were selected and optimized appropriately.
MD simulations obtained the spatial aggregation morphology of three asphaltene molecules. Their shear viscosity was calculated by non-equilibrium molecular dynamics (NEMD) simulations to study the relationship among asphaltene molecular structure, viscosity, and aggregation behavior. Finally, the weak intermolecular interactions and their stability in the process of asphaltene aggregation were visualized by aRDG and TFI methods.
Molecular model
In this work, the simulation study of model asphaltene was based on the structure of actual asphaltene molecules characterized by Schuler27 using AFM and STM in 2017. We selected three types of asphaltene molecules with different arrangements and appropriate optimization, referred to as AsphalteneO, AsphalteneT, and AsphalteneY. The molecular structures are shown in Fig. 2(a). The cluster-like benzene ring structures are formed by several benzene rings, and the chain-like structures are formed by stacks of several benzene rings. AsphalteneO has a cluster-like aromatic ring structure, AsphalteneY has a branched chain-like aromatic ring structure, and AsphalteneT has both cluster-like and branched chain-like benzene ring structures.
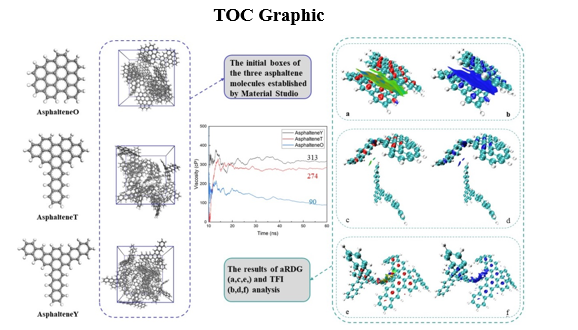
In addition, the initial boxes of the three asphaltene molecules were established by Material Studio, as shown in Fig. 2 (b). Each box contained a random distribution of 30 asphaltene molecules with an initial density of 0.6 g/cm3 for later NEMD simulation analysis.
NEMD Method
Non-equilibrium molecular dynamics (NEMD) has been employed to study the shear viscosity of fluids through various techniques28, 29. The NEMD calculation imposes shear boundary conditions on the system of interest and is expected to drive it to a steady state. The dynamic behavior of the interatomic forces in the NEMD simulation was calculated by using the isothermal atomic formula of the SLLOD Eq. 30–32. The velocity Verlet algorithm was used to solve the numerical integration of the velocities and positions of the particles33. In the NEMD calculation, the constant shear rate γ was applied to the infinite periodic array of the subsystem of volume V, and each subsystem contained N particles28. The shear viscosity \(\eta\) was calculated using the following formula:
$$\eta =–\frac{{P}_{\alpha \beta }}{\gamma }$$
1
where \(\eta\) is the viscosity, \(\gamma\) is the shear rate, and \({P}_{\alpha \beta }(\alpha ,\beta =x,y)\) is the pressure component of the system in Cartesian coordinates in the shear field,
$${P}_{\alpha \beta }=\frac{1}{V}\left(\sum _{i}{m}_{i}{v}_{i}^{\alpha }{v}_{i}^{\beta }+\sum _{i}\sum _{j>i}{r}_{ij}^{\alpha }{F}_{ij}^{\beta }\right)$$
2
\({m}_{i}\) is the mass of the ith particle, \({\nu }_{i}\) is the relative velocity of the ith particle, \({r}_{ij}\) is the distance between particles \(i\) and \(j\), and \({F}_{ij}\) is the force between particles \(i\) and \(j\).
Material Studio software was only used for modeling, and all MD simulations were performed with the large-scale atomic/molecular massively parallel simulator (LAMMPS) program package. The specific steps were as follows:
A. A box with 30 randomly distributed asphaltene molecules was created by Material Studio, and the initial density of the system was set to 0.6 g/cm3. This study used the COMPASS force field.
B. The density and energy of the randomly generated simulation box (including the kinetic and potential energy evaluation) were calculated. Each system was balanced by an NPT ensemble simulation under the constant pressure of 1 atm for 10 ns, and the temperature was kept at 333 K, which defined the correct density for the system and box size.
C. The NVT simulation was carried out for 50 ns at 333 K using the Nose-Hoover thermostat. The shear rate was 1E-7/fs, the time step was 1 fs, and periodic boundary conditions were applied in all directions. (Under a high strain rate, the sharp increase in pressure prevented the NEMD method from calculating asphaltene viscosity34, so the shear rate selected in this study was 1E-7/fs.)
D. After 10 ns, the atomic displacement, force, velocity, potential energy, kinetic energy, viscosity, and other parameters related to the complete relaxation and equilibration of the system were recorded for subsequent analysis.
Averaged Reduced Density Gradient
Reduced Density Gradient (RDG)
To reveal the weak interactions, Johnson et al.20 constructed the formula based on the study of reduced density gradient (RDG) as a function of electron density ρ(r):
$$\text{R}\text{D}\text{G}\left(\text{r}\right)=\frac{1}{2{\left(3{\pi }^{2}\right)}^{1/3}}\frac{\left|\nabla \rho \left(r\right)\right|}{{\rho \left(r\right)}^{4/3}}$$
3
where \(▽\) is the gradient operator, ρ (r) is the electron density,\(|▽\rho \left(r\right) |\) is the norm of the electron density gradient.
RDG, a method to characterize and visualize different types of weak interactions, defines an actual space function that enables its value to distinguish between regions with different characteristics in the system. Suppose a particular weak interaction has a larger ρ(r) at its critical point. In that case, ρ(r) will generally be larger in the surrounding region due to the continuity of ρ(r). Therefore, the strength of the interaction is apparent at a glance by mapping the numerical magnitude of ρ(r) to the RDG equivalent surface in different colors. However, ρ(r) can only reflect the strength of the interaction, and the type of interaction needs to be reflected by the sign (λ2) function (where λ2 is the second largest eigenvalue of the Hessian matrix of electron density). The electron density ρ(r) and the sign (λ2) function are combined to obtain the sign (λ2) ρ function projected onto the RDG equivalent surface, which reveals the location, strength, and type of the weak interaction. The color scale is set to blue, green, and red. The blue region exhibits more potent, attracting weak interactions, and the green region has a small ρ(r), indicating weak interaction strength. The van der Waals interaction region fits this feature. The red region corresponds to the stronger region of the potential barrier effect (also known as nonbonded overlap) in the benzene ring. Specific applications and analyses are discussed in detail in the Results and discussion section.
Averaged Reduced Density Gradient (aRDG)
The aRDG method is very similar to the RDG method in principle. Like the RDG method, the aRDG method shows the weak interaction region through the equivalent surface of the RDG function. It projects the sign (λ2) ρ function on the RDG equivalent surface with different colors to show the weak interaction types. However, in the aRDG method, the electron density, the gradient of the electron density, and the Hessian matrix of the electron density are used to calculate the RDG and sign (λ 2) ρ functions, which are all obtained by averaging over the multi-frame structure. Therefore, both the RDG function and the sign (λ2) ρ function in the aRDG method represents the average RDG and the average sign (λ2) ρ for the whole trajectory. The calculation formulas are as follows:

different regions was stable.
The asphaltene molecular trajectory file was obtained in this study by NEMD simulation. Then the asphaltene molecular interaction and its stability were analyzed by aRDG and TFI using Multiwfn software35 developed by the Lu Tian team. In this process, pro-molecular density21 built-in Multiwfn software was applied to reduce the calculation time. The results of the aRDG and TFI analysis in this study can be displayed by the VMD program36.
Shear Viscosity
Figure 4 shows the viscosity changes of the three kinds of asphaltene molecules during the NEMD simulations. The average viscosity data were calculated every 10 steps, 50 steps, 100 steps, and 1000 steps. The viscosities obtained at the four frequencies converged utterly, and the convergence values were almost the same. The total time of the simulation process was 60 ns. Due to the excessive fluctuations in the data during the first 10 ns, statistical analysis was carried out using the data collected after 10 ns. Finally, the viscosity changes of three asphaltenes during 10–60 ns were obtained. The viscosity began to converge after 55 ns. According to the convergence, the viscosity of asphaltene was obtained. The viscosities of AsphalteneY, AsphalteneT, and AsphalteneO were 313 cP, 274 cP, and 90 cP, respectively, indicating that the viscosities of asphaltene molecules are closely related to their structure.
Asphaltene molecules with different structures have different aggregation mechanisms. Therefore, to further understand the viscosity of asphaltene, the aggregation behavior of asphaltene is analyzed in the following section, which provides conditions for explaining the differences in the molecular viscosities of the asphaltene molecules with different structures.
Asphaltene aggregation
In this study, the three asphaltene molecules are classified as AsphalteneO, AsphalteneT, and AsphalteneY according to their structure. VMD visually analyzed the three asphaltene molecules after the NEMD simulations to study the specific aggregation modes with different structures, as shown in Fig. 5. Most of the AsphalteneO molecules aggregated via face-to-face stacking, and a few molecules underwent face-to-face stacking and offset stacking. AsphalteneT has both cluster-like benzene ring and chain-like benzene ring structures; the cluster-like benzene ring structures of AsphalteneT molecules stack face-to-face, but the chain-like benzene ring structures are staggered, which is classified as offset stacking in this study. In addition, some AsphalteneT molecules form T-shaped aggregates where the cluster-like benzene ring structure of one molecule is in contact with that of another molecule, and the two molecules are distributed vertically with an overall T-shape. On the other hand, AsphalteneY forms mainly T-shaped or cross-shaped aggregates related to the molecular structure. Some molecules associate via offset stacking because they contain chain-like benzene ring structures.
According to the previous section, the viscosities of the three asphaltene molecules are in the order AsphalteneY > AsphalteneT > AsphalteneO, which can be fully explained by the aggregation behaviors in this section. AsphalteneO tends to undergo face-to-face stacking under the conjugation effect of cluster-like benzene ring structures. AsphalteneO molecules move relatively smoothly under the action of a shear field. However, in addition to offset stacking, AsphalteneY undergoes T-shaped cross stacking between molecules, which appears to hinder the movement of the molecules structurally. AsphalteneT has the conjugation effect of cluster-like benzene ring structures and the structural hindrance effect of chain-like branched benzene ring structures. In other words, different aggregation methods lead to different viscosities. Asphaltene viscosity is a macroscopic expression of asphaltene molecule aggregation, and asphaltene molecules showing T-shaped stacking aggregation have higher viscosities (AsphalteneY and AsphalteneT), while asphaltene molecules showing face-to-face stacking aggregation have lower viscosities (AsphalteneO).
Fig. 6 shows the variation process of the number of aggregates of the three asphaltenes during the simulation. Comparing Fig. 6 (a), (b) and (c), we can conclude that the number of asphaltene aggregates in the box is relatively stable and varies in a certain number interval. During the simulation, the number of aggregates of the three kinds of asphaltenes constantly varies, especially between 10 and 60 ns, which may be caused by the rupture and reorganization of asphaltene aggregates under shear. Additionally, the viscosity of asphaltene is influenced by the aggregation and disruption processes. The viscosity reflects the fluid's internal resistance, which is related to the rupture and reorganization of the aggregates. Perhaps high resistance corresponds to slow rupture and reorganization, and low resistance corresponds to fast rupture and reorganization. As seen in Fig. 6 (a) and (b), Asphaltene O and Asphaltene T have a low number of aggregates and a relatively large overall fluctuation. In contrast, Asphaltene Y has a higher number of aggregates and relatively smooth variation, which explains the different viscosity sizes of the three asphaltenes. As for some intermittent jaggedness appearing in Fig. 6 (a), (b) and (c), it is due to the abrupt change in the number of asphaltene aggregates within a short time or short step, and then it returns to a stable level.
Moreover, we can guess that the interactions between Asphaltene Y molecules should be larger and more complex than those of Asphaltene O, making their rupture recombination behavior under the shear field different. Therefore, we will next use aRDG analysis to reveal the intermolecular interactions of the three asphaltenes and provide data to support this conjecture.
aRDG analysis
The asphaltene molecules' intermolecular interaction types and stabilities can be directly visualized by using aRDG and TFI analysis and software for MD simulations and visualization. In the aRDG analysis, we used the following colors to identify the types of interactions: blue indicates strong attractive interactions, such as hydrogen bonds; green represents weak interactions, such as van der Waals interactions; and red indicates repulsive interactions, such as steric hindrance, as shown in Fig. 8(k). In the TFI analysis, blue was used to describe interactions that were almost unaffected and highly stable when undergoing thermal motion, red indicated interactions that were easy to destroy and unstable during thermal motion, and green was used to indicate interactions that fluctuated between the "blue" and "red" types. The spikes on the left and right sides of the scatter diagram (Fig. 7) represent the attractive and spatial repulsive forces between the two molecules, respectively. The results of aRDG and TFI analyses are shown in Fig. 8.
The large green area in Fig. 8(a) shows a wide range of van der Waals interactions between AsphalteneO molecules, and the corresponding spike-tip x value on the scatter diagram is approximately ± 0.015, as shown in Fig. 7(a). There is a red spindle region in the middle of the benzene ring, showing a strong steric effect, which corresponds to the spike on the far right of the scatter diagram. Notably, there is an area between C-H and C-H, half of which is red and half blue, corresponding to spikes with x values of approximately + 0.03 and − 0.03, respectively. This isosurface shows both a steric effect and an attraction effect, and mutual exclusion and attraction effects coexist. However, there is no strong attraction between C-H and C-H. This part experiences only van der Waals interactions because aRDG is based on the excimer density approximation, so the electron density in some weak interaction regions is not sufficiently realistic and may be overly high. According to the analysis of the TFI in the figure (blue area in Fig. 8(b), the weak interaction between AsphalteneO molecules has little influence on thermal motion and is relatively stable in the system.
On the one hand, Fig. 8(a) and (c) show that the weak interactions between AsphalteneT and AsphalteneO molecules are very similar. On the other hand, the van der Waals forces between the T-shaped stacked AsphalteneT molecules, such as the two small green areas shown in Fig. 8(e), are weaker. The spike corresponding to the scatter plot is thinner than that of the offset-stacked AsphalteneT molecules, as shown in Fig. 7(b) and (c).
Compared with the interactions of AsphalteneT and AsphalteneO, the weak intermolecular interactions of offset-stacked AsphalteneYs are slightly more complicated; AsphalteneY molecules do not have strong van der Waals interactions. Figure 8(g) shows that green and blue merge on the equivalent surface of AsphalteneY. For another stacking method of AsphalteneY, as shown in Fig. 8(i), the equivalent surfaces between the two molecules are green and blue. There are van der Waals interactions between the chain-like structures of benzene rings. However, compared with the other two kinds of asphaltene molecules, AsphalteneY has more blue isosurfaces and stronger attractions, which is one of the most significant reasons it has the highest viscosity in this study. Meanwhile, the results of the analysis verify the conjecture in the previous section
Additionally, according to the TFI analysis, the weak intermolecular interactions of the three kinds of asphaltenes are relatively stable and slightly affected by thermal motion, as shown in Fig. 8(b), (d), (f), (h) and (j).
This study selected three representative asphaltene molecules (AsphalteneO, AsphalteneT, and AsphalteneY) to investigate the relationship among the weak intermolecular interaction of asphaltene molecules, viscosity, and aggregation behavior using NEMD simulations and the aRDG method. The results indicated that the viscosity of asphaltene molecules was related to their intermolecular interactions, and the order of the viscosity values was AsphalteneY > AsphalteneT > AsphalteneO. The reason for this is that the weak intermolecular interactions of AsphalteneY are slightly more complex compared to AsphalteneT and AsphalteneO, including Stronger attractive forces. The weak interactions between AsphalteneT molecules are similar to those between AsphalteneO molecules, mainly van der Waals interactions and steric hindrance effects. On the other hand, the difference in asphaltene molecular structure leads to different aggregation modes, which affects the movement of asphaltene under the action of a shear field. In addition, TFI analysis was used to visualize the weak intermolecular interactions and their stability for asphaltene molecules. The weak intermolecular interactions of the three asphaltene molecules have good stability and are slightly affected by thermal motion.
The asphaltene molecular models employed in this study are relatively simple (containing only aromatic rings). At the same time, alkyl chains and heteroatoms (O, S, N) are also essential factors that affect the viscosity, aggregation behavior, and other properties of asphaltenes. Therefore, in the future, we intend to investigate the possible effects of alkyl chain length and heteroatoms on the viscosity and aggregation behavior of asphaltenes. We plan to employ the aRDG method to study the weak intermolecular interactions of asphaltene molecules with different lengths of alkyl chains and with and without the addition of heteroatoms.
Conflicts of interest/Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethics approval
The manuscript is prepared in compliance with the Ethics in Publishing Policy as described in the Guide for Authors.
Consent to participate
The manuscript is approved by all authors for publication.
Consent for publication
The consent for publication was obtained from all participants.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Code availability
The calculations have been carried out using LAMMPS and Materials Studio 2017 R2.
Acknowledgments
This work was funded by Young Projects of the Hunan Provincial Education Department (18B066), Hunan Provincial Natural Science Foundation of China (2022JJ30562), Fundamental Research Funds for the Central Universities (2020kfyXJJS127), Open Research Fund Program of Science and Technology on Aerospace Chemical Power Laboratory (STACPL220181B05), and Basic Research Project (XXXX-2019-083). The authors are sincerely grateful for their support. The views and findings of this study represent those of the authors and may not reflect those agencies.
Author contributions
Qunchao Lin: writing - original draft, conceptualization, methodology, and software; Lei Deng: writing-review & editing, validation, and formal analysis; Ge Dong: writing - original draft, conceptualization, methodology, and software; Xianqiong Tang: data curation, visualisation and funding acquisition; Wei Li: conceptualization, funding acquisition, and writing - review& editing; Zhengwu Long: software, methodology, and writing - review & editing; Lingyun You: supervision, conceptualization, methodology, formal analysis, and writing - review & editing; Fu Xu: supervision, resources, conceptualization, and writing - review & editing. Qunchao Lin and Ge Dong contributed equally.
- Sedghi M, Goual L, Welch W, et al (2013) Effect of asphaltene structure on association and aggregation using molecular dynamics. J Phys Chem B 117:5765-5776. https://doi.org/10.1021/jp401584u
- Wang H, Xu H, Jia W, et al (2017) Revealing the Intermolecular Interactions of Asphaltene Dimers by Quantum Chemical Calculations. Energy & Fuels 31:2488-2495. https://doi.org/10.1021/acs.energyfuels.6b02738
- You L, Spyriouni T, Dai Q, et al (2020) Understanding of Structural and Surface Tension Properties of Asphalt Model Using Molecular Dynamics Simulation. RILEM International Symposium on Bituminous Materials.
- Luo P, Gu Y (2007) Effects of asphaltene content on the heavy oil viscosity at different temperatures. Fuel 86: 1069-1078. https://doi.org/10.1016/j.fuel.2006.10.017
- You L, Spyriouni T, Dai Q, et al (2020) Experimental and molecular dynamics simulation study on thermal, transport, and rheological properties of asphalt. Construction and Building Materials 265: 120358. https://doi.org/10.1016/j.conbuildmat.2020.120358
- Silva H S, Sodero A C R, Bouyssiere B, et al (2016) Molecular Dynamics Study of Nanoaggregation in Asphaltene Mixtures: Effects of the N, O, and S Heteroatoms. Energy & Fuels 30: 5656-5664. https://doi.org/10.1021/acs.energyfuels.6b01170
- Long Z, You L, Tang X, et al (2020) Analysis of interfacial adhesion properties of nano-silica modified asphalt mixtures using molecular dynamics simulation. Construction and Building Materials 255: 119354. https://doi.org/10.1016/j.conbuildmat.2020.119354
- Mullins O C, Sabbah H, Eyssautier J, et al (2012) Advances in Asphaltene Science and the Yen–Mullins Model. Energy & Fuels 26: 3986-4003. https://doi.org/10.1021/ef300185p
- Carauta A N M, Correia J C G, Seidl P R, et al (2005) Conformational search and dimerization study of average structures of asphaltenes. Journal of Molecular Structure: THEOCHEM 755: 1-8. https://doi.org/10.1016/j.theochem.2005.02.063
- Sjoblom J, Simon S, Xu Z (2015) Model molecules mimicking asphaltenes. Adv Colloid Interface Sci 218: 1-16. https://doi.org/10.1016/j.cis.2015.01.002
- Hunter C A, Sanders J K M (1990) The nature of .pi.-.pi. interactions. Journal of the American Chemical Society 112: 5525-5534. https://doi.org/10.1021/ja00170a016
- Pacheco-Sánchez J H, Zaragoza I P, Martínez-Magadán J M (2003) Asphaltene Aggregation under Vacuum at Different Temperatures by Molecular Dynamics. Energy & Fuels 17: 1346-1355. https://doi.org/10.1021/ef020226i
- Takanohashi T, Sato S, Tanaka R (2004) Structural Relaxation Behaviors of Three Different Asphaltenes Using MD Calculations. Petroleum Science and Technology 22: 901-914. https://doi.org/10.1081/LFT-120038716
- Rogel E (2000) Simulation of Interactions in Asphaltene Aggregates. Energy & Fuels 14: 566-574. https://doi.org/10.1021/ef990166p
- He L, Wang P, He L, et al (2018) Molecular dynamics simulations of the self-organization of side-chain decorated polyaromatic conjugation molecules: phase separated lamellar and columnar structures and dispersion behaviors in toluene solvent. RSC Advances 8:11134-11144. https://doi.org/10.1039/c7ra13101a
- Jian C, Tang T, Bhattacharjee S (2013) Probing the Effect of Side-Chain Length on the Aggregation of a Model Asphaltene Using Molecular Dynamics Simulations. Energy & Fuels 27: 2057-2067. https://doi.org/10.1021/ef400097h
- Long Z, Tang X, Ding Y, et al (2022) Influence of sea salt on the interfacial adhesion of bitumen–aggregate systems by molecular dynamics simulation. Construction and Building Materials 336: 127471.https://doi.org/10.1016/j.conbuildmat.2022.127471
- Schuler B, Meyer G, Pena D, et al (2015) Unraveling the Molecular Structures of Asphaltenes by Atomic Force Microscopy.J Am Chem Soc 137 :9870-6. https://doi.org/10.1021/jacs.5b04056
- Ekramipooya A, Valadi F M, Farisabadi A, et al (2021) Effect of the heteroatom presence in different positions of the model asphaltene structure on the self-aggregation: MD and DFT study. Journal of Molecular Liquids 334: 116109. https://doi.org/10.1016/j.molliq.2021.116109
- Johnson E R, Keinan S, Mori-Sanchez P, et al (2010) Revealing noncovalent interactions. J Am Chem Soc 132: 6498-506. https://doi.org/10.1021/ja100936w
- Wu P, Chaudret R, Hu X, et al (2013) Noncovalent Interaction Analysis in Fluctuating Environments. Journal of Chemical Theory and Computation 9: 2226-2234. https://doi.org/10.1021/ct4001087
- Wu W, Wang C, Xu H, et al (2020) Study of the aggregation behaviour of three primary reactive dyes via molecular dynamics simulations. Molecular Simulation 46: 627-637. https://doi.org/10.1080/08927022.2020.1755037
- Bai Q, Yao X (2016) Investigation of allosteric modulation mechanism of metabotropic glutamate receptor 1 by molecular dynamics simulations, free energy and weak interaction analysis. Sci Rep 6: 21763.https://doi.org/10.1038/srep21763
- Li M-R, Zhang N, Zhang F-S (2018) A simulation study of water property changes using geometrical alteration in SPC/E. Chinese Physics B 27: 083103. https://doi.org/10.1088/1674-1056/27/8/083103
- Lv W, Xu G, Zhang H, et al (2015) Interlayer water regulates the bio-nano interface of a beta-sheet protein stacking on graphene. Sci Rep 5: 7572. https://doi.org/10.1038/srep07572
- Zhang J, Zhang L, Xu Y, et al (2018) Deciphering the binding behavior of flavonoids to the cyclin dependent kinase 6/cyclin D complex. PLoS One 13: e0196651. https://doi.org/10.1371/journal.pone.0196651
- Schuler B, Fatayer S, Meyer G, et al (2017) Heavy Oil Based Mixtures of Different Origins and Treatments Studied by Atomic Force Microscopy. Energy & Fuels 31: 6856-6861. https://doi.org/10.1021/acs.energyfuels.7b00805
- Berker A, Chynoweth S, Klomp U. C, et al (1992) Non-equilibrium molecular dynamics (NEMD) simulations and the rheological properties of liquid n-hexadecane. Journal of the Chemical Society, Faraday Transactions 88: 1719-1725.. https://doi.org/10.1039/FT9928801719
- Long Z, You L, Xu, F, et al (2022) Nanomechanical-atomistic insights on interface interactions in asphalt mixtures with various chloride ion erosion statuses. J Colloid Interface Sci 628: 891-909. https://doi.org/10.1016/j.jcis.2022.08.014
- Pan G, Ely J F, Mccabe C, et al (2005) Operator splitting algorithm for isokinetic SLLOD molecular dynamics. The Journal of Chemical Physics 122: 094114. https://doi.org/10.1063/1.1858861
- Mundy C J, Siepmann J I, Klein M L (1995) Decane under shear: A molecular dynamics study using reversible NVT‐SLLOD and NPT‐SLLOD algorithms. The Journal of Chemical Physics 103: 10192-10200. https://doi.org/10.1063/1.469922
- Separdar L, Bailey N P, Schrøder T B, et al (2013) Isomorph invariance of Couette shear flows simulated by the SLLOD equations of motion. The Journal of Chemical Physics 138. https://doi.org/10.1063/1.4799273
- Martys N S, Mountain R D (1999) Velocity Verlet algorithm for dissipative-particle-dynamics-based models of suspensions. Physical Review E 59: 3733. https://doi.org/10.1103/PhysRevE.59.3733
- Ding Y, Huang B, Shu X (2018) Modeling Shear Viscosity of Asphalt through Nonequilibrium Molecular Dynamics Simulation. Transportation Research Record: Journal of the Transportation Research Board 2672: 235-243. https://doi.org/10.1177/0361198118793316
- Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33: 580-592. https://doi.org/10.1002/jcc.22885
- Guixa-Gonzalez R, Rodriguez-Espigares I, Ramirez-Anguita J M, et al (2014) MEMBPLUGIN: studying membrane complexity in VMD. Bioinformatics 30: 1478-1480. https://doi.org/10.1093/bioinformatics/btu037
No competing interests reported.
{kind=link}