AIL identified from TCM monomers is a novel potential inhibitor targeting c-Jun
To develop c-Jun inhibitors, we focused on TCM monomers, the treasure trove of bioactive compounds, and screened out antitumor drug candidates with anti-inflammatory effects due to the rapid development of immunotherapy in the treatment of melanoma. 114 compounds (Additional file 1: Table S1) relevant to both tumor and inflammation were selected from 1,616 TCM monomers according to the literature, and then, we processed 114 compounds and c-Jun by MOE software (2016 version) as well as conducted molecular docking in induced fit mode. Finally, the 25 top compounds were initially screened out as potential inhibitors targeting c-Jun (Additional file 2: Fig. S1A). To obtain reliable c-Jun inhibitors, a ligand-based QSAR integration model was employed to further screen for the activity of inhibitors. Then, we collected a dataset of known binding small molecules of c-Jun from ChEMBL and BindingDB. Previous study proved that the consensus QSAR model constructed by CATS, MACCS and MOE2D, which performance is significantly better than a single model [28]. Therefore, we also adopted random forest (RF) to build a consensus QSAR model based on 5-fold cross-validation. Our results showed high prediction accuracy of the consensus QSAR model (Additional file 1: Table S2). With a value of 0.5 as the threshold, 6 compounds with predictive values greater than 0.5 were screened for further verification (Additional file 1: Table S3). Furthermore, the pharmacokinetics and toxicity properties of 6 potential inhibitors were calculated through ADMETlab (Additional file 1: Table S4). Among those candidates, Britannin (BRT) and AIL were finally identified as candidate inhibitors with high lipid solubility and water solubility, high bioavailability, low cardiotoxicity, and low hepatotoxicity (Additional file 1: Table S4 and Fig. 1B).
AIL is the main active constituent extracted from the bark of Ailanthus altissima (Fig. 1C). To further validate that AIL was a targeted inhibitor of c-Jun, we performed a combinatorial target prediction strategy for the stepwise screening of the potential targets of AIL (Additional file 2: Fig. S2A). First, four ligand-based prediction platforms, including Swiss TargetPrediction, ChEMBL, Similarity Ensemble Approach (SEA) and PPB2, were employed to narrow the target space of the query molecule AIL [29], and we obtained 26 targets in total. Subsequently, we found that these targets were closely related to cancer and inflammation according to the top enriched KEGG pathways (Additional file 2: Fig. S2B), suggesting that the functions of preliminary screening targets were consistent with the phenotypic effects of AIL. To acquire reliable prediction, a Venn diagram was then plotted to observe the overlay of targets predicted by at least three prediction tools (Additional file 2: Fig. S2C). Finally, 5 molecules, including proto-oncogene c-Jun (JUN), androgen receptor (AR), 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), glucocorticoid receptor (NR3C1) and protein kinase C alpha type (PRKCA) (Fig. 1D), were identified as potential targets of AIL.
We also applied a consensus QSAR model to further screen the key targets of AIL. The evaluation metrics accuracy (ACC) and area under the curve (AUC) were all above 0.7 (Fig. 1E-F), indicating the good predictive ability of the QSAR models. The prediction results based on the consensus QSAR model were outputted as probability values and are listed in Fig. 1D. Here, with a probability value of 0.5 as the default cut-off, the proto-oncogene c-Jun and AR were selected as potential targets. A previous study reported that AIL downregulated AR protein levels [30]. Interestingly, our prediction showed that the c-Jun score was higher than the AR score (Fig. 1D), suggesting that our prediction was relatively reliable and that c-Jun was most likely to be the target of AIL. To further validate our hypothesis, molecular docking between c-Jun and AIL was performed to observe their binding. As shown in Fig. 1G, the main binding force is hydrogen binding; therefore, we confirmed the interaction between AIL and c-Jun.
AIL suppresses melanoma progression in vitro and in vivo
To study the role of AIL in melanoma, we examined the viability of the normal epidermal melanocytes (PIG1), the mouse epidermal JB6 cells, the human melanoma cell lines SK-MEL-5 and SK-MEL-28, and the mouse malignant melanoma cell lines B16-F10 and YUMM1.7 treated with AIL by CCK-8 assay. As shown in Fig. 2A and Additional file 2: Fig. S3A, AIL inhibited cell proliferation with IC50 values (48 h) of 2.039 µmol/L in PIG1 cells, but inhibited cell proliferation with IC50 values (48 h) of 0.3725, 0.1255, 0.2245 and 0.6663 µmol/L in SK-MEL-5, SK-MEL-28, A-375 and WM35 cells, respectively. In addition, AIL inhibition of IC50 values (48 h) was calculated to be 14.99 µmol/L in JB6 cells, but inhibited cell proliferation with IC50 values (48 h) of 1.214 and 3.301 µmol/L in B16-F10 and YUMM1.7 cells, respectively (Fig. 2B), indicating that melanoma cells are more sensitive to AIL treatment than the normal cells. We next explored the effects of AIL on metastasis and invasion of melanoma cells by wound healing assay and transwell invasion assay. As shown in Fig. 2C-D and Additional file 2: Fig. S3B-C, AIL significantly inhibited the metastasis and invasion of the human and mouse melanoma cell lines. To further validate the antitumor effects of AIL, we inoculated B16-F10 mouse melanoma cells into C57BL/6 mice for an in vivo study. As shown in Fig. 2E-G, AIL treatment significantly reduced tumor volume and weight compared with the control group, and both 0.5 and 5 mg/kg AIL application did not cause a significant difference in mouse body weight among tumor-bearing mice (Fig. 2H). Together, these results suggest that AIL serves as a promising therapeutic candidate in the treatment of melanoma.
AIL binds to c-Jun and induces its degradation
Next, to verify the direct interaction between AIL and c-Jun in melanoma cells, we collected SK-MEL-5, SK-MEL-28, B16-F10 and YUMM1.7 cell lysates and incubated them with AIL-Sepharose 4B beads. The results showed that c-Jun bound to the AIL-Sepharose 4B beads complex but not the Sepharose 4B beads alone (Fig. 3A and Additional file 2: Fig. S4A). To further test whether c-Jun was a target of AIL, we detected c-Jun expression in PIG1, SK-MEL-5, SK-MEL-28, JB6, B16-F10 and YUMM1.7 cells. Compared with non-malignant cells, c-Jun was highly expressed in melanoma cells (Fig. 3B). As expected, after transfecting c-Jun into PIG1 and JB6 cells, cell viability was enhanced (Additional file 2: Fig. S4B); however, these cells were more sensitive to AIL treatment than the control cells (Fig. 3C-D). To investigate the role of AIL in c-Jun, we detected the c-Jun mRNA and protein levels after AIL treatment in melanoma cells. As shown in Fig. 3E-G and Additional file 2: Fig. S4C-E, AIL had no significant effect on c-Jun transcription (Fig. 3E and Additional file 2: Fig. S4C) but reduced c-Jun protein expression in a dose-dependent and time-dependent manner in melanoma cells (Fig. 3F-G and Additional file 2: Fig. S4D-E). We also observed that AIL treatment resulted in a significant downregulation of c-Jun in tumor tissues compared with the control (Additional file 2: Fig. S4F). Since c-Jun is a highly unstable protein because of polyubiquitination [31], we wondered whether c-Jun was regulated by the proteasome pathway in AIL-treated melanoma cells. Intuitively, the half-life of c-Jun was shortened after AIL treatment (Fig. 3H and Additional file 2: Fig. S4G), and we also observed that MG-132, a proteasome inhibitor, could rescue the downregulation of c-Jun in melanoma cells after AIL treatment (Fig. 3I and Additional file 2: Fig. S4H). Thus, we provide evidence here that AIL targets c-Jun by reducing its protein stability.
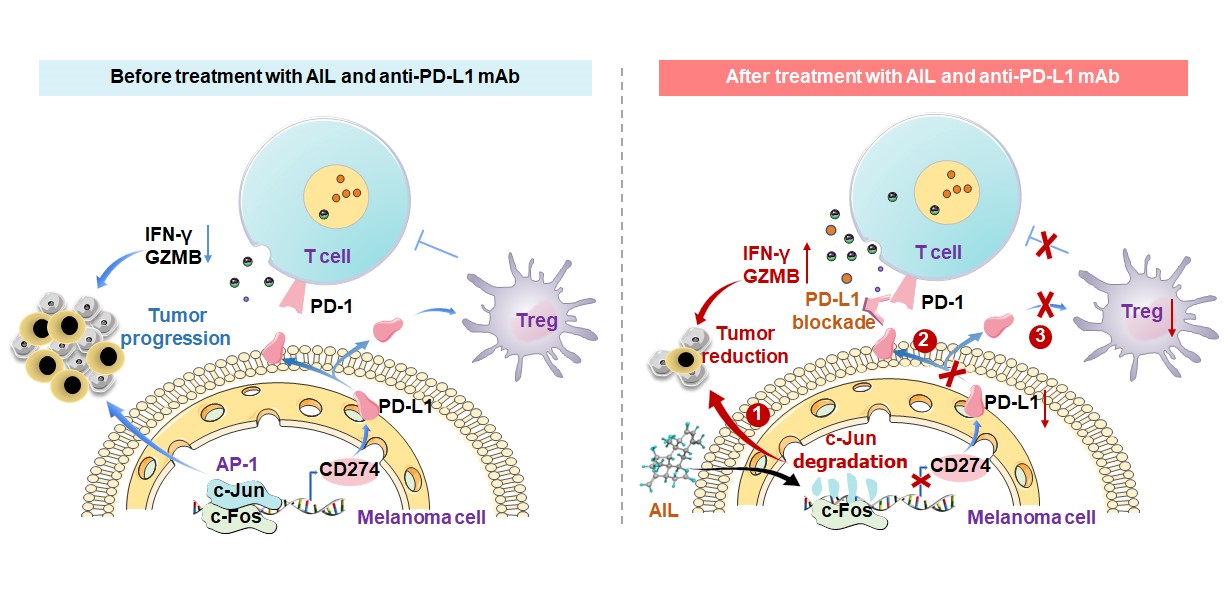
AIL improves the therapeutic efficacy of PD-L1 blockade by inhibiting the infiltration of Tregs
Combination therapy is a promising strategy to improve the efficacy of anti-PD-L1 mAb. To validate the effect of AIL treatment on immunotherapy, we utilized an anti-PD-L1 mAb to treat immune-competent mice inoculated with B16-F10 melanoma cells. We treated B16-F10 tumor-bearing mice with PBS plus IgG isotype CTRL (IgG2a), AIL plus IgG2a, PBS plus anti-PD-L1 mAb or AIL plus anti-PD-L1 mAb. Anti-PD-L1 treatment was performed every 3 days, and AIL treatment was performed every 2 days (Fig. 4A). We captured tumors on the ninth day, and tumor growth was measured every 2 days (Fig. 4A). In the B16-F10 tumor-bearing mice, AIL or anti-PD-L1 mAb treatment alone both reduced mouse tumor burden (Fig. 4B). More importantly, cotreatment with AIL and anti-PD-L1 mAb further decreased xenograft tumor volume and weight (Fig. 4C and Additional file 2: Fig. S5A). We observed that AIL, anti-PD-L1 mAb and combination treatments of AIL and anti-PD-L1 mAb had no significant difference compared with the CTRL group in terms of mouse body weight (Additional file 2: Fig. S5B). At the end of the treatment, tumor samples were harvested for further flow cytometry analysis. Interestingly, combined treatment with AIL and anti-PD-L1 mAb resulted in a significantly lower number of Tregs than each treatment alone (Fig. 4D). We also found that both AIL and anti-PD-L1 mAb increased the number of CD4 + T cells and CD8 + T cells, but the effect of combination therapy on CD4 + T cells and CD8 + T cells was not different from that of AIL or anti-PD-L1 mAb treatment alone (Additional file 2: Fig. S5C). Furthermore, we investigated the production of IFN-γ and granzyme B (GZMB) by CD4 + T cells and CD8 + T cells. As shown in Fig. 4E-H, the populations of IFN-γ + CD8 + T cells, GZMB + CD8 + T cells, IFN-γ + CD4 + T cells and GZMB + CD4 + T cells were significantly increased in both the AIL group and the anti-PD-L1 mAb group and synergistically improved by the combination treatment. We also detected the change in macrophages (F4/80 + CD11b+) and the percentage of Gr-1 + CD11b + MDSCs in CD45 + cells. We noticed an increase in the M1 (F4/80 + CD11b + MHC class II+) macrophage population in both the AIL treatment group and the anti-PD-L1 mAb group (Additional file 2: Fig. S5D). Additionally, AIL and anti-PD-L1 mAb treatment substantially reduced the infiltration of M2 (F4/80 + CD11b + CD206+) macrophages and MDSCs (Additional file 2: Fig. S5E and F). However, when AIL and anti-PD-L1 mAb treatment were combined, changes in M1 macrophages, M2 macrophages and MDSCs were no more significant than when AIL or PD-L1 was treated alone (Additional file 2: Fig. S5D-F). Compared to the control group, CD45-PD-L1 + cells showed a significant decrease upon AIL or anti-PD-L1 mAb treatment, which was further suppressed by combination treatment with AIL and anti-PD-L1 mAb (Fig. 4I), indicating that PD-L1 expression was distinctly inhibited in tumors. However, there were no significant differences detected in the populations of NK1.1 + cells, NK1.1 + PD-L1 + cells, CD4 + CTLA4 + cells, and CD8 + CTLA4 + cells (Additional file 2: Fig. S5G-J), suggesting that the immune effects of AIL and the anti-PD-L1 mAb were independent of these cells. Taken together, these results support the concept that AIL and anti-PD-L1 mAb treatment substantially reduce the Tregs and improve the cytotoxicity of T cells.
AIL inhibits PD-L1 transcriptional expression by regulating c-Jun
Previous results showed a reduction in PD-L1 expression after treatment with AIL (Fig. 4I). To identify the effect underlying AIL-mediated regulation of tumor PD-L1 levels, we detected the mRNA level of PD-L1 after AIL treatment in different melanoma cell lines. As shown in Fig. 5A and Additional file 2: Fig. S6A, AIL decreased PD-L1 mRNA levels in a dose-dependent manner. We also found that AIL significantly inhibited PD-L1 levels in the supernatant (Fig. 5B). Furthermore, western blot analysis showed that the expression of PD-L1 decreased in a dose- and time-dependent manner in cells treated with AIL (Fig. 5C-D and Additional file 2: Fig. S6B-C). Previous studies have shown that c-Jun, the target of AIL, is a common TF mediating the transcription of many tumor-relevant genes, such as cyclin D1, p53, and INK4A [32–34]. We thus speculated that c-Jun was also a potential regulator of PD-L1 in melanoma cell lines. To verify our hypothesis, we identified the potential sequences in the promoter region of PD-L1 recognized by c-Jun (-1,200 bp~-1 bp) through the PROMO database (Fig. 5E) and constructed a PD-L1 luciferase reporter gene. The results of the luciferase assay showed that overexpression of c-Jun significantly enhanced PD-L1 promoter activity, which could be blocked by AIL treatment (Fig. 5F). In addition, we performed a ChIP and confirmed that c-Jun could directly bind to the promoter of PD-L1 and transcriptionally regulate PD-L1 mRNA levels, but AIL treatment inhibited their interaction (Fig. 5G-H and Additional file 2: Fig. S6D). To determine the downregulation of PD-L1 expression under AIL treatment through c-Jun inhibition, we overexpressed c-Jun in SK-MEL-5, SK-MEL-28, B16-F10 and YUMM1.7 melanoma cell lines (Fig. 5I and Additional file 2: Fig. S6E). We observed that overexpression of c-Jun significantly increased the PD-L1 mRNA and protein levels compared to the control group (Fig. 5K and Additional file 2: Fig. S6F-I). In addition, overexpression of c-Jun partially rescued the downregulation of PD-L1 caused by AIL (Fig. 5K and Additional file 2: Fig. S6F-I). However, overexpression of c-Jun actually resulted in more significant inhibition of PD-L1 by AIL (Fig. 5J and Additional file 2: Fig. S6G-H). Taken together, our results support the hypothesis that AIL treatment reduces PD-L1 expression via the AIL-c-Jun-PD-L1 axis.
AIL attenuates Treg differentiation through the c-Jun/PD-L1 axis
In the TME, Tregs contribute to tumor immune escape by strongly inhibiting T-lymphocyte immunity [20]. The above results indicated that AIL blocked Treg-mediated immunosuppression and enhanced the cytotoxicity of T cells. To further verify the effect of AIL on Tregs, we isolated naive CD4 + T cells from the spleen of C57BL/6 mice. Additional file 2: Fig. S7B shows that the efficiency of naive CD4 + T-cell selection reached 97.3%. Then, naive CD4 + T cells were cultured with AIL-treated tumor supernatant under Treg-prone conditions for 3 days (Fig. 6A). As shown in Fig. 6B-C, the percentage of Tregs was reduced following AIL-treated tumor supernatant treatment. Moreover, we further assessed the mRNA level of Foxp3 (a vital regulatory molecule expressed in Tregs that regulates the unique genetic features and transmission regulatory activity of Tregs [24]). In accordance with the flow cytometry results, AIL-treated tumor supernatant also inhibited the transcription of Foxp3 (Fig. 6D-E). Additionally, we assessed the effects of AIL on Treg differentiation (Additional file 2: Fig. S7A). AIL treatment also caused mild inhibition of Treg differentiation and Foxp3 mRNA levels (Additional file 2: Fig. S7C-E). A previous study showed that c-Jun was a TF of Foxp3 [35], indicating that the inhibitory effect of AIL on Tregs may be mediated by the AIL-c-Jun-Foxp3 axis. We then compared the effects of AIL and AIL-treated tumor supernatant on Treg differentiation. Interestingly, AIL-treated tumor supernatant exhibited more significant inhibitory effects on Treg differentiation and Foxp3 mRNA levels (Additional file 2: Fig. S7C-E), suggesting that the inhibitory effect of AIL on Treg differentiation is mainly determined by molecules in the tumor supernatant. It was previously reported that PD-L1 beads alone could convert naive CD4 + T cells to Foxp3 + Tregs in vitro [24]. We speculated that AIL might suppress the secretion of PD-L1 by targeting c-Jun, thus blocking Treg differentiation and further promoting the cytotoxicity of T cells. To confirm our hypothesis, we examined the effects of tumor supernatant on Treg differentiation after c-Jun overexpression in melanoma cells (Additional file 2: Fig. S7F). As expected, overexpression of c-Jun significantly promoted Treg differentiation (Additional file 2: Fig. S7G, H) and elevated Foxp3 mRNA levels (Additional file 2: Fig. S7I, J). In addition, overexpression of c-Jun partially rescued the Treg differentiation inhibition (Additional file 2: Fig. S7G-H) and Foxp3 downregulation (Additional file 2: Fig. S7I-J) caused by AIL. More importantly, our results showed that AIL-treated tumor supernatant inhibited Treg differentiation (Fig. 6F-G) and Foxp3 expression (Fig. 6H-I) more significantly after c-Jun overexpression, further suggesting that AIL indeed works by targeting c-Jun. Taken together, these results indicate that AIL inhibited Treg differentiation through the AIL-c-Jun-PD-L1 axis.
{kind=link}