AKI caused by renal I/R often occurs in the clinic and is associated with high mortality, resulting in patients experiencing shock and massive blood loss, which requires kidney surgery (21, 22). AKI induced by I/R accounts for approximately 60% of total AKI cases, especially in Intensive Care Unit (ICU) patients (22). To reduce the mortality caused by I/R, the development of new effective strategies to prevent and treat local and remote organ injury following renal I/R is crucial for improving the prognosis of patients. Numerous pharmacological models and substances have been studied, but none have been completely successful (23–25). Following kidney ischemia, in the process of restoring blood flow, the first injury event is that mitochondria produce a large amount of ROS. In addition, the increase of non-mitochondrial reactive oxygen species leads to secondary tissue damage and subsequent inflammation. Therefore, both mitochondrial and non-mitochondrial reactive oxygen species play a critical role in ischemia-reperfusion injury (26). ROS production during reperfusion is crucial in I/R-induced AKI, and as such, inhibiting the formation of ROS may serve as a potential treatment for ischemic AKI.
In this study, we found that after I/R-induced AKI mice were intraperitoneally injected with Rb1, renal damage was reduced, blood creatinine and urea nitrogen levels were also decreased, the inflammatory response was downregulated (Fig. 1), and the mitochondrial function of HK-2 cells was improved (Fig. 3), suggesting that Rb1 may be useful as a new treatment of I/R-induced AKI. Rb1 is a kind of dammarane triterpenoid saponin, which is considered to be one of the most important active components in ginseng and a primary component playing a pharmacological role. Rb1 has many beneficial effects on the human body, including cardiovascular and central nervous system activities, anti-diabetic, and anti-tumor activities (27–30). Rb1 can reduce oxidative stress, inhibit inflammation, downregulate MDA, inhibit ROS, and promote GSH expression, due to its beneficial characteristics.
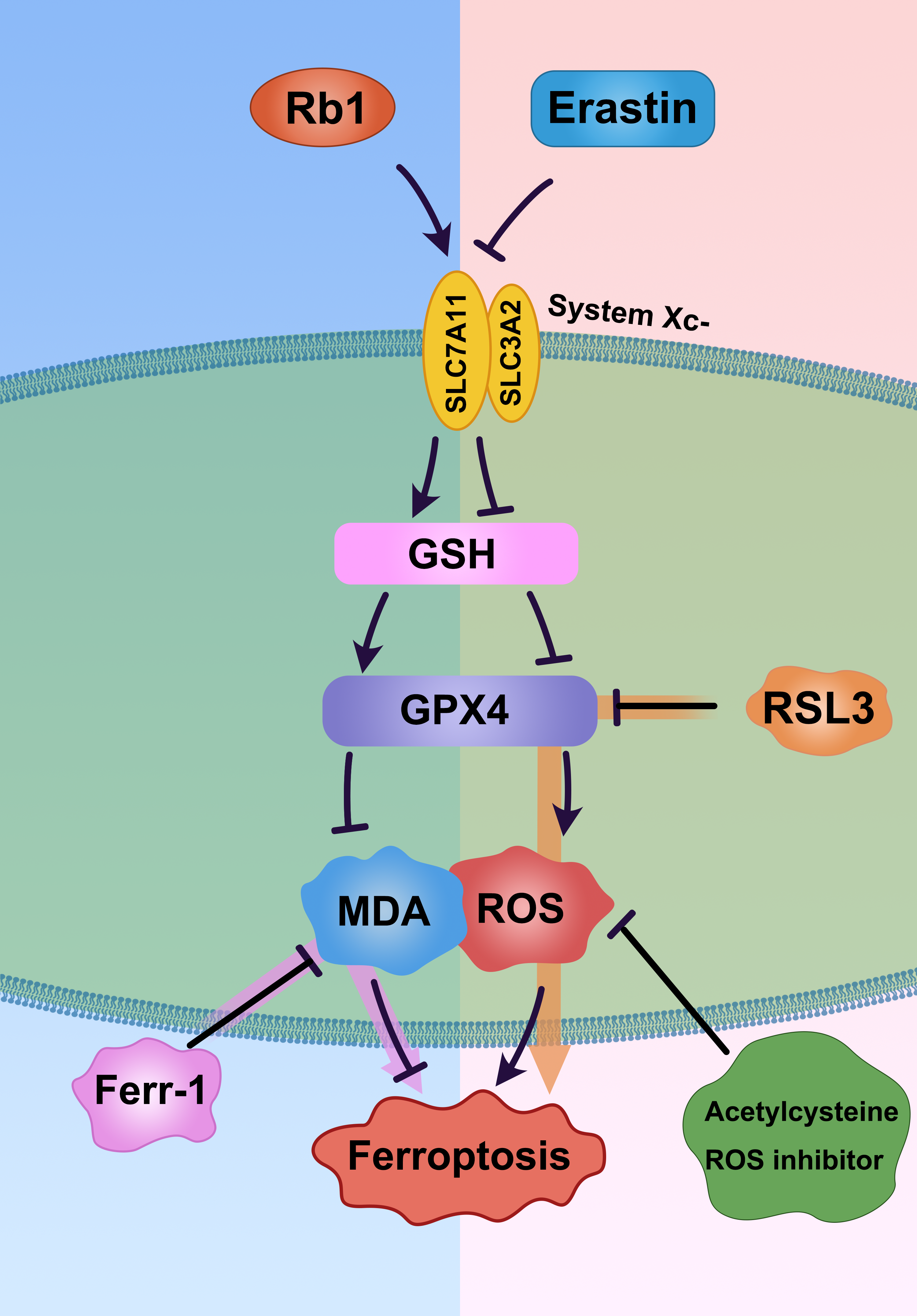
ROS is involved in damaging organizations in many ways. Mitochondria are the primary source and target of reactive oxygen species. Excessive ROS causes oxidative stress and mitochondrial dysfunction (31). Ferroptosis is non-apoptotic programmed cell death, and its primary performance is that under the action of iron or ester oxygenase, it catalyzes the unsaturated fatty acids highly expressed on the cell membrane to produce liposome peroxidation, thereby inducing cell death. In addition, ferroptosis is also manifested in the reduction of the expression of antioxidant systems (glutathione GSH and glutathione peroxidase-4: GPX4) (13, 32). GPX4 is a phospholipid hydroperoxidase, which plays a key role in protecting cells from membrane lipid peroxidation. Inactivation of GPX4 leads to ferroptosis and stimulates the production of activated oxygen (33). ROS formation induced by GPX4 inactivation is considered to be the executor of ferroptosis (34). SLC7A11 is the primary subunit of the Xc- system, which transports cystine into cells for the synthesis of GSH. Therefore, inhibiting the expression of SLC7A11 can induce ferroptosis. As a tumor suppressor gene, p53 enhances the sensitivity of cells to ferroptosis by downregulating the expression of the Xc- system component SLC7A11. ROS specifically induces p53-mediated ferroptosis, with the level of SLC7A11 being crucial to this process (13).
Rb1 was determined to reduce myocardial I/R injury by inhibiting the production of ROS by mitochondrial complex I. In addition, ROS-induced inflammatory response, mitochondrial dysfunction, and endoplasmic reticulum stress also promote apoptosis, with all these ROS-related signaling pathways ultimately promoting the occurrence of AKI (31). In sepsis, Rb1 significantly reduces LPS or cantharidin-induced acute kidney injury and saves LPS-induced sepsis mice from death (35). In diabetes nephropathy, Rb1 inhibits the excessive production of ROS mediated by aldose reductase (AR) and has a protective effect on mitochondrial damage induced by high glucose, effectively alleviating the progress of diabetes nephropathy (18). Rb1 intervention can reduce the apoptosis of renal tubular epithelial cells (36). In chronic kidney disease, Rb1 can effectively reduce the degree of oxidative stress and inflammation in patients with CKD by inhibiting ROS, as well as slowing down the progress of CKD in the early stage (37, 38). However, the role of Rb1 in I/R-induced AKI has not been reported.
In this study, we confirmed for the first time that Rb1 can reduce AKI induced by I/R in vivo. Taking Rb1 as an example, we further confirmed that Rb1 protected AKI induced by I/R by inhibiting ferroptosis (Fig. 7). We further confirmed our hypothesis through in vitro experiments, through which we determined that Rb1 reduces ferroptosis and reactive oxygen species formation induced by the inactivation of GPX4 and SLC7A11 (Fig. 7). Erastin (an SLC7A11 inhibitor) binds and inactivates SLC7A11, mediating SLC7A11-dependent ferroptosis (13). RSL3 (a GPX4 inhibitor) binds and inactivates GPX4, mediating GPX4-dependent ferroptosis (34). RSL3 and Erastin reduced the expression of GPX4 and SLC7A11 in HK-2 cells, significantly upregulated the expression of ROS and MDA; increased the expression of ferrous ions; and downregulated the expression of GSH (Fig. 3), which promotes the occurrence of iron death. When Rb1 co-cultured HK-2 cells with RSL3 or Erastin, the results indicated that ROS and MDA models were downregulated, GSH was significantly upregulated, ferrous ions in cells were reduced, ferroptosis in HK-2 cells was significantly reduced, and cell activity was increased (Fig. 3). Further, ferrostatin-1 was used to co-culture HK-2 cells with RSL3 or Erastin. The results demonstrated that ferrostatin-1 could simulate the protective effect of Rb1 on HK-2 cells (Fig. 5). We further confirmed that Rb1 reduced HK-2 cell damage by interfering with ferroptosis. In addition, the co-culture of HK-2 cells with ROS inhibitor (acetylcysteine) and RSL3 or Erastin showcased that the intracellular ferrous ion and ROS were downregulated, and the cell activity was significantly increased.
In conclusion, our results confirm that Rb1 promotes GSH production through antioxidation, upregulates the expression of GPX4 and SLC7A11, inhibits ROS, and depresses ferroptosis in renal tubular epithelial cells (Fig. 7). Previous studies have found that ferroptosis and oxidative stress play a key role in renal ischemia-reperfusion injury (39). Our findings may expand the scope of Rb1 in the kidney and provide new drug targets for the treatment of acute renal injury.
{kind=link}