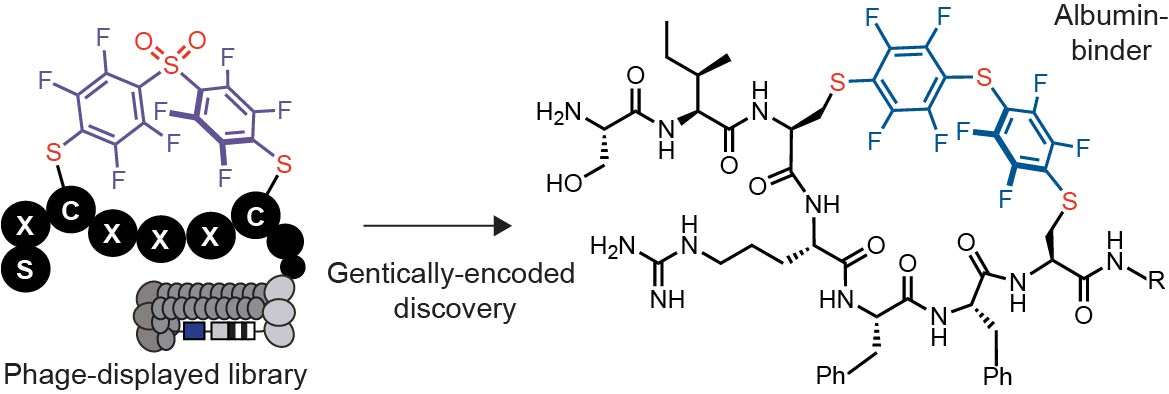
Selection of albumin binders
We devised and conducted three discovery campaigns that used different library architecture and selection strategies. In the first discovery campaign, we modified the phage libraries of structure SXCX4–5C with DFS following a previously published protocol and confirmed that 85% of the phage library is modified to yield octafluoro-diphenylsulfone-crosslinked macrocycles (OFS-SXCX4–5C-phage) (Figure 2A, Figure S1A).42
We performed three rounds of phage selection using HSA coated to the surface of 96 well polystyrene plates as bait. In parallel, we screened the same library on polystyrene wells coated with Protein A (negative control) to distinguish specific HSA-binding sequences from poly-specific protein binding sequences (Figure S1A). In round 3, the phage recovery of the OFS-macrocycle library selection against HSA two-fold increase compared to round 1 and round 2 but only a minor increase compared to selection against unrelated protein (Figure S1D). The recovery of unmodified round 3-library panned against HSA was 17-fold lower than the recovery of the OFS-macrocycle library, indicating that the OFS linchpin contributes to protein binding (Figure S1D). Differential enrichment (DE) analysis of the next-generation sequencing (NGS) of all test and control experiments (Table S1) identified several families of peptide macrocycles that had statistically significantly higher (p<0.05) enrichment in binding to HSA when compared to binding to unrelated protein (Figure S1 B–C). The analysis yielded three consensus motifs: STCHDITC (1a), STCHYIGC (2a) and STCHANC (3a) (Figure S1E).
The second discovery campaign employed HSA immobilized on a 96 well plate in rounds 1 and 3, and biotinylated HSA as bait immobilized onto streptavidin beads in round 2 (Figure S2A). In round 3, the phage recovery of the OFS-macrocycle library selection against HSA increased by a factor of 200 when compared to round 1 and round 2. The recovery of the unmodified library panned against HSA was insignificant (Figure S2D). The binding of the OFS-macrocycle phage library recovered from round 3 to Protein A, ConA and Casein was 2, 14, and 300-fold lower respectively when compared to recovery on HSA-coated wells (Figure S3). These observations suggested that (i) specific albumin-binding sequences had been selected, and (ii) the binding of these sequences to albumin required presence of OFS linchpin (Figure S3). A DE analysis of NGS data (Figure S4, Table S2) identified sequences that were significantly (p<0.05) enriched in the screen against HSA but not control proteins. The LOGO analysis yielded a consensus motif: STCHTIYC (4a) (Figure S2E). Although the original libraries were designed as SXCXnC where n=4 and 5, they contained a small fraction of SXCX3C sequences,44 and we observed the enrichment of such sequences in the selection. To explore the apparent preference for smaller macrocycles, we devised a third selection campaign that employed only SXCX3C libraries modified with DFS (Figure 2A).
The small diversity of the library made it possible to employ a single round of panning and NGS-DE analysis and to identify the binders. To mimic the complex serum environment, the panning was conducted using a mixture of biotinylated HSA (Bio-HSA), His-tag fusion T4-PG protein (His6-T4-PG) and unlabelled milk proteins as bait. In a control selection, we used the same mixture with biotinylated ConA (Bio-ConA) in place of Bio-HSA (Figure 2B). Proteins were captured with streptavidin or Ni-NTA affinity beads, respectively. The captured phage DNA was liberated from beads by treatment with hexane and the released DNA was amplified by PCR (Figure S4) and sequenced with Illumina deep sequencing (Figure 2B, Table S3). A DE analysis identified a set of 85 sequences that were significantly enriched (p<0.05, >3-fold) in the screen against Bio-HSA when compared to the screen against His6-T4-GP and Bio-ConA (Figure 3A–B, Figure S5). We applied a pairwise amino acid clustering to identify the 85 hit sequences (Figure 3C) and observed 8 motifs: FF, MF, MG, TK, GM, PV, VY and KR associated with these enriched sequences (Figure 3D). Based on this analysis, we nominated sequences SICRFFC (5a), SFCPMFC (6a) and SLCKREC (7a) as hits and STCQGEC (8a) as a negative control for chemical synthesis, and further validation (Figure 3E).
Validation of albumin binders
We observed non-specific reactivity of OFS-macrocyclic peptides with thiol nucleophiles such as glutathione (GSH) over several hours in basic pH (Figure S6). Replacing DFS with a less reactive pentafluorophenyl sulfide (Figure 1D) abolished the undesired reactivity: The resulting perfluorophenylsulfide (PFS)-macrocycles were unreactive to 2-mercaptoethanol over three weeks and unreactive towards free thiol on HSA (Figure S7). Molecular dynamics simulation suggested the OFS-macrocycles and the PFS-macrocycles exhibit similar ground state conformational landscape (Figure S8). Many perfluoro-aryl crosslinked macrocycles were poorly soluble in water, and we synthesized them with either a GGKKK or GGG tag at the C-terminus to increase their solubility; some sequences were synthesized with both tags to check whether these affect HSA binding. The C-terminal tags aided in providing sufficient solubility properties for downstream analyses (Figure S9).
The unique fluorine handle in perfluoro-aryl modified peptides made it possible to determine their binding to HSA using 19F NMR (Figure 4). In a typical experiment, we maintained peptide concentration at 50uM and HSA at 100uM (Table S4). We observed broadening of and disappearance of 19F signals that correspond to fluoroaromatic groups, which indicated the binding of the peptide to HSA (Figure 4A, Figure S10). We could not fit a definitive Kd value to the binding response due to the complex binding behaviour and quality of the NMR signal. However, in an albumin titration series, one can use qualitative estimates such as the concentration of albumin necessary to suppress 50% of the initial fluorine signal. Based on these qualitative analyses, it was apparent that some peptides (e.g., PFS-SICRFFCGGG) have stronger binding to HSA, whereas other macrocycles (e.g., PFS-STCQGECGGG) have weaker binding towards HSA (Figure 4A, Figure S10). By measuring the decrease in the signal at a fixed concentration of peptide and HSA, we evaluated 8 sequences found in all discovery campaigns (Figure 4B, S11) and we nominated PFS-SICRFFCGGG (5c) as the “hit” and PFS-STCQGECGGG (8c) as the negative control for further investigation. Peptides modified at the C-terminus with either GGKKK or GGG solubility tags have similar binding affinity (Figure 4B, Figure S11). We titrated 5c and 8a against rat serum albumin and observed similar binding to rat and human albumin (Figure S12). We attempted to confirm the binding affinity of these sequences by isothermal titration calorimetry (ITC) using SA-21 as a control;37 however, a complex multi-site binding behaviour for all peptides obscured the accurate evaluation of binding affinity by ITC (Figure S13–15). The 19F NMR assay, thus, was critically enabling for validation and ranking of the albumin binding leads.
A fluorescence polarization binding assay (FP) successfully measured the binding affinities of the macrocycles with the fluorophore BODIPY at the C- or N-terminus. In a typical experiment, we used PFS-SICRFFCGGG (5c) or PFS-SFCPMFCGGG (6c) at 1 µM concentration and titrated HSA from 0.1 µM to 100 µM. The dose-response curve could be fit to a single-state binding model with binding affinity of Kd = 4–6 µM for 5c and at least 100 times weaker affinity for 6c (Figure 5A and S16–S19). BODIPY alone bound weakly to HSA with > 300 µM binding affinity (Figure 5A and S16–S19). The FP-assay made it possible to measure binding to other proteins or even complex mixtures (serum). A titration of the mouse serum (Figure S18) yielded a similar binding profile to that observed in binding to pure albumin (Figure 5A). Replacing HSA with lysozyme and RNAse, the assay detected no binding response, confirming that 5c binding was specific to HSA (Figure 5B).
Switching location of the fluorescent probe from the N-terminus to C-terminus did not significantly change the affinity of 5c (Kd = 4-6 µM, Figure S19). The switching from DFS to PFS also exhibited a minimal effect on the binding of peptides 5b and 5c (Figure S20). The results from FP were in the same order of magnitude as the semi-qualitative estimates acquired for BODIPY-free peptides by the 19F NMR binding assay, indicating that the presence of a fluorophore did not significantly increase the binding (Figure S21). Heinis and co-workers recently observed that fluorophores could dramatically increase binding affinity for albumin, and removing the fluorophore is detrimental to the binding of the albumin binder.21 To exclude this possibility, we conducted an NMR-binding assay of N-terminally-labelled PFS-SICRFFCGG and BODIPY-free peptides. We observed that the binding affinity was similar (Figure S21).
Elucidation of the binding pocket for perfluoro-macrocycles
We evaluated whether binding pockets of 5c are similar to known albumin binders: carbamazepine, diclofenac and ibuprofen (Figure 6A). We observed that the binding of PFS-SICRFFCGGG (5c) did not decrease in the presence of any of these drugs; thus, it does not share the same binding pocket as carbamazepine, diclofenac, or ibuprofen (Figure 6B). To follow on this observation, we performed a series of docking calculations to seek the most favorable binding locations of PFS-SICRFFCGGG (5c) on the surface of HSA.
Nine distinct sites on HSA were previously shown to bind to fatty acids,45 some of which also bind to other ligands such as ibuprofen and diclofenac46, 47 (Figure S22). These nine reported fatty acid binding sites on five different initial HSA structures were selected for docking of 5c. Figure 7A shows the HSA protein with some of its bound fatty acids, based on the pdbID 1e7e45. Overlaid with this structure is 5c docked to the corresponding fatty acid binding sites on the HSA surface. Figure 7B shows the binding scores for 5c–HSA complexes across different HSA structures, based on the distinct pdbIDs and different binding site locations (complete results summarized in Figure S23 and Table S5). Consistently, 5c has the most favorable binding score in binding site 1, with the value of –8.95 ± 1.0 kcal/mol, averaged over all the docking calculations performed. Therefore, the results in Figure 7B suggest that the primary HSA binding site for 5c is binding site 1. The next most favorable binding sites are sites 8, 6, and 7, with the most favorable binding scores of –6.6 ± 1.1 kcal/mol, –6.2 ± 0.7 kcal/mol, and –6.0 ± 1.2 kcal/mol, respectively (Figure 7, Table S5). We observed binding sites 1 and 8 are near to each other on the HSA surface, with the center of mass distance between fatty acids occupying these sites being 5.3 Å. As such, it is unlikely that binding sites 1 and 8 can be simultaneously occupied by two 5c molecules. Figure 7A shows the four HSA residues that interact with the fatty acid in binding site 1 via charge and nonpolar interactions. In contrast, PFS-SICRFFCGGG (5c) has more interactions with this HSA binding site, including the HSA residues R114, R117, Y138, Y161, I142, L154, S193. Notably, HSA residues R117, Y138, and Y161 in binding site 1 are found to mediate HSA interactions with both the fatty acid and 5c.
Combined docking results (Figure 7) and binding observation (Figure 6) suggested that ibuprofen, diclofenac and 5c bind to different locations on the HSA surface. The structural studies46 demonstrated that ibuprofen binds to the binding sites labeled by 3/4 and 6 (pdbID 2bxg, Figure S22), which are distant from the HSA binding site 1. Structure of HSA bound to diclofenac48 (pdbID 4z69, Figure S22) contains two HSA chains in it. One of the HSA chains has a single diclofenac at the binding site 7, while the second HSA chain has three bound diclofenac ligands in total, with two also located at the binding site 7, and the third located near the binding site 1, which is also occupied by a bound fatty acid. The structure locations suggest that diclofenac has the strongest binding to binding site 7, since it is observed there in both HSA chains, and a weaker binding to binding site 1, as only one single HSA chain is observed with diclofenac nearby.
Circulation lifetime of albumin peptides in mice
To evaluate the half-life circulation of albumin-binding perfluoro-macrocycles, we injected a mixture of peptides PFS-SICRFFCGG (5c), weak binding peptide PFS-STCQGECCGGG (8c) as the negative control and SA-21 as the positive control into mice and monitored the remaining peptide level by LC–MS (Figure 8A, Figure S24).
We observed that the negative control 8c disappeared below the limit of detection after 5 min (Figure 8B). The concentration of 5c decreased 10-fold and SA-21 concentration decreased 5-fold after 2 hours. The combined results confirm a significant retention of PFS-SICRFFCGG peptide in circulation when compared to unrelated macrocyclic peptides with minimal to no detectable binding to HSA. The single digit micromolar peptide does not rival the mid-nanomolar SA-21 peptide, and the observed differences in half-life likely reflect the relative affinities for albumin. The PFS-SICRFFCGG peptide, thus, provides an attractive minimalistic starting point for further attenuation of binding affinity for albumin and subsequent attenuation of circulation half-life.
{kind=link}