DNA methylation is the most comprehensively investigated human epigenetic mark, and mQTLs have been widely investigated in disease-relevant adult tissues to fine map causal variants and effector genes at genetic risk loci in complex diseases. To date, the investigation of mQTLs during human development has been limited (18,29–31), and studies have predominantly focused upon cerebral tissues and neuropsychiatric disorders. Such analyses have not been previously conducted in the developing human skeleton. In this report, we used a total of 100 human embryonic and fetal skeletal tissue samples to identify differences in DNAm between developmental and aged limbs and investigate the existence of OA genetic risk mechanisms during development.
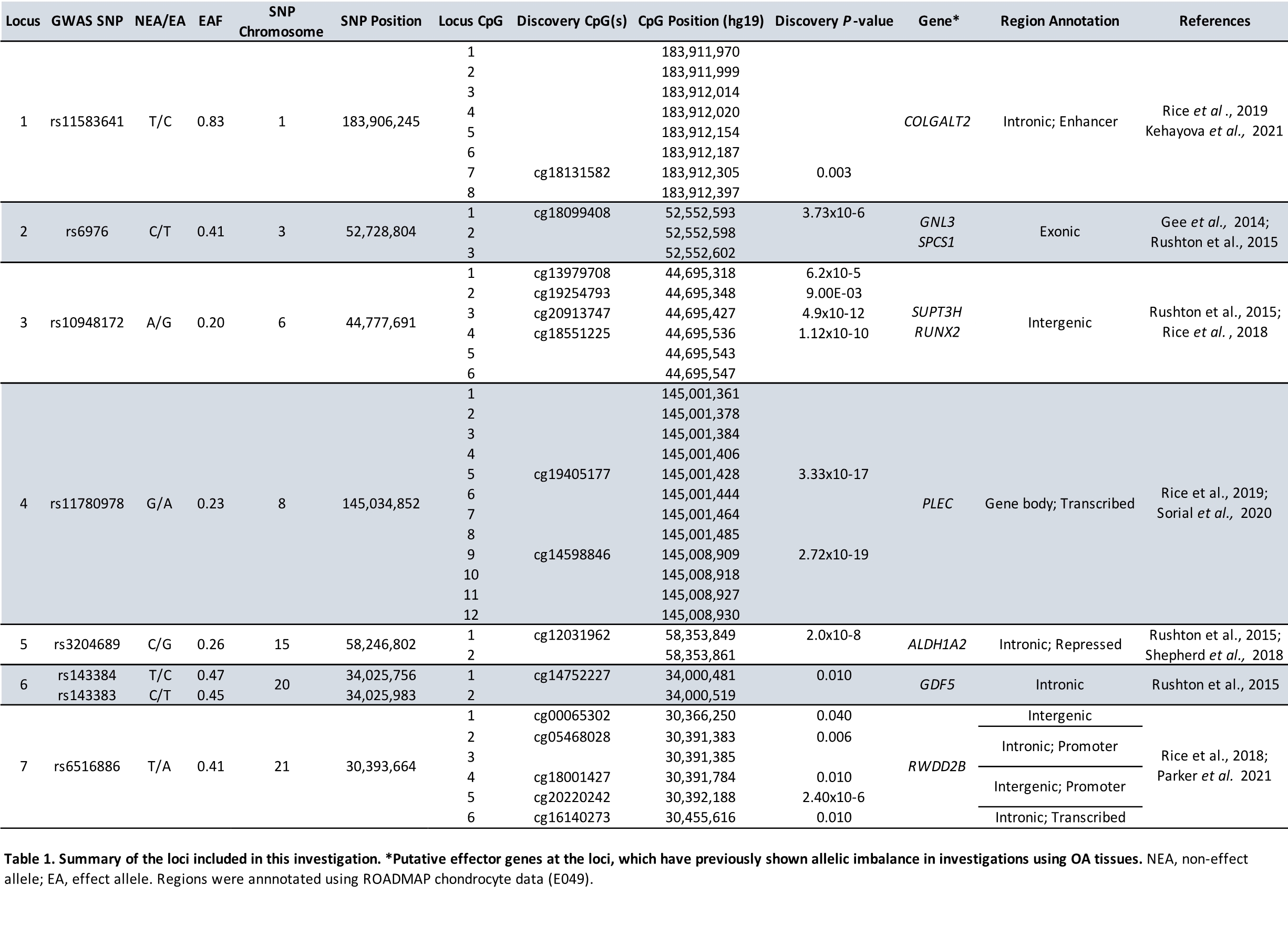
We investigated 39 CpGs: sites of, or adjacent to, well-characterized OA-mQTLs in aged cartilage. We identified significant DMS at 67% of CpGs when comparing developmental and aged cartilage, the majority of which became hypermethylated in aged cartilage. This is noteworthy, as in ageing tissues a trend towards global hypomethylation has been observed, known as the epigenetic drift. The relationship between DNAm and age is complex, however the consensus is that DNAm increases across tissues in the early years of life, and then gradually declines with age due to a loss in maintenance stringency (32,33). Furthermore, the co-methylation of proximal CpGs has also been observed to decline with older age (34). We identified co-methylation clustering of CpGs at the seven investigated loci. At Locus 4 (PLEC) we investigated two distal (7.5kb) clusters of CpGs (21) at which DNAm was co-regulated, with strong negative correlations between the CpGs, reflecting the 3D architecture of DNA (35). Conversely, at Locus 7, we investigated 6 mQTL CpGs, only three of which showed a significant co-regulation from the SNV. Importantly, the co-methylation did not appear to be lost with age at any of the loci, suggesting that the epigenetic effects captured by this targeted study are directly relevant to CpGs that are tightly regulated by SNVs and involved in pathogenesis of tissue-specific diseases.
We identified OA-mQTLs at 28% of investigated CpGs in FL tissues, and at 85% in FC. We attribute the relative depletion of mQTLs in the FL samples to both a relatively small sample size (n = 19), coupled with mQTL tissue specificity. Whilst this is difficult to disentangle at loci where smaller effect sizes were observed, the absence of FL mQTLs at Locus 4, harboring PLEC, can be attributed to a cartilage-specific effect (CpG 9 GE 71.9% in FC, 6.7% in FL).
One of the most striking observations that we made during this study, was the inconsistency in the pattern of observations between the loci, which indicates that the inter-locus mechanisms behind the observed epigenetic changes are distinct. From this we hypothesize that the functional impacts of these risk loci are exerted at different spatiotemporal points during the human life course. An epigenome wide study would potentially allow the identification of clusters of loci that show comparable effects and share regulatory timepoints. Yet, in the current targeted study, this is not possible to determine.
Analyses of transcript expression at the nine OA effector genes identified significant AEI at 6/9 genes (P < 0.016) in FC, confirming the presence of differential allelic gene expression. This verifies that the functional genetic risk of OA is occurring at the majority of the investigated loci during skeletal development. We investigated meQTLs to determine whether this was being driven by the differential DNAm and identified significant correlations at 15/39 CpGs: FL (Locus 2 and 4), FC (Locus 7), and AC (Locus 1, 2, 4). Yet, the inability to widely detect linear correlations between DNAm and the expression of a potential target gene is perhaps unsurprising. The question of DNAm as a cause vs. consequence has long been debated, and the answer has been hindered due to the complex involvement of multiple cis-regulatory elements (CREs) in the finetuning of gene expression. A recent study used novel single-cell technologies in mouse embryonic stem cells to investigate the co-occurrence of DNAm with chromatin accessibility, and TF occupancy (36). The authors demonstrated that at most enhancers, DNAm does not antagonize chromatin accessibility, nor the binding of TFs. However, they identified a subset of cell-type specific enhancers at which DNAm directly regulates TF binding, and target gene expression (36). We postulate that such a subset of methylation-sensitive enhancers would be enriched in targeted mQTL investigations such as ours, where the CREs in which they fall have prior association with disease. This is supported by our recent research using dCas9 epigenome modulators, which have shown a direct causality between DNAm and gene expression at OA loci (22,23,25). The identification of tissue specific methylation sensitive enhancers in OA would be bolstered by the expansion of molecular epigenetic investigations to include additional tissues of the articulating joint, rather than solely focusing on cartilage; the inclusion of joint tissues from young adults, although their acquisition in sufficient numbers will be challenging; and the adoption of modern single-cell technologies to look for subpopulations of cells driving disease etiology within the joint.
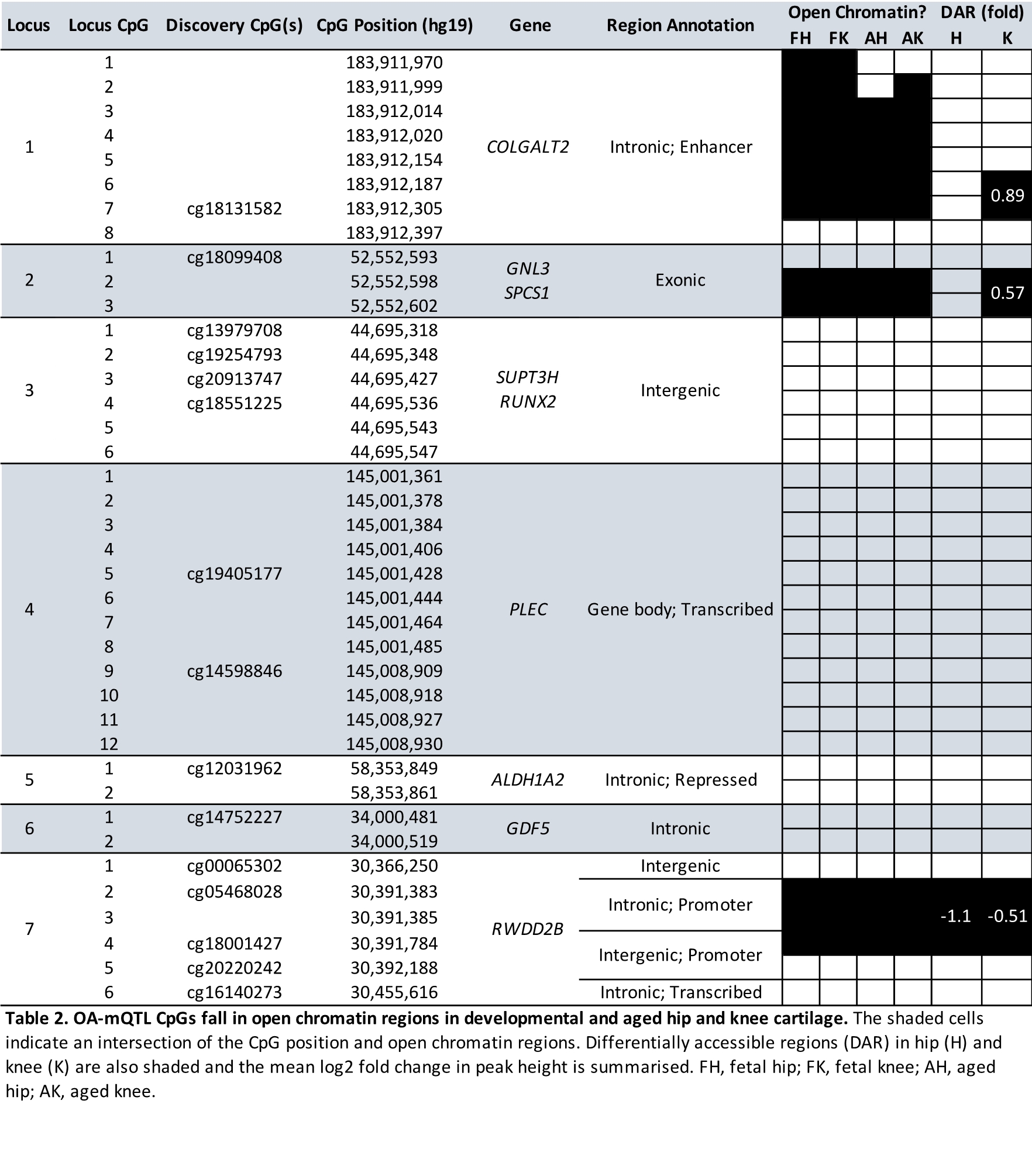
Finally, we conducted ATAC sequencing and identified open chromatin regions in developmental and aged samples. The identified accessible regions in the fetal cartilage were enriched for TF binding motifs including SOX9, the master regulatory TF essential for chondrogenesis (37), and FOS/JUN, TFs that can form homo- and heterodimers essential in cartilage development (38–40). Intriguingly, this motif was also enriched in the aged cartilage accessible regions. The FOS/JUN transcription factors have also been shown to play a role in OA pathogenesis (41), and our data further substantiates the notion that stringent regulation of the binding of these TFs to their motifs is integral to cartilage health and function throughout the human life course. We were further able to prioritize causal SNVs across the seven loci, and our data identified three loci at which the investigated CpGs fell into open chromatin regions.
At Locus 1, 8 CpGs were investigated across a COLGALT2 enhancer (22). Generally, levels of DNAm were significantly lower and the genotypic effect stronger in the developmental cartilage, consistent with our observation of increased chromatin accessibility during development. Yet, whilst all 7 mQTL CpGs were differentially methylated between FC and AC, the difference in mean DNAm at CpG6 was just 4.5%. Additionally, CpG6 was relatively hypomethylated in the AC samples, when compared to the rest of the investigated sites, and this was the site of the highest measured GE at the locus (55.7%). Furthermore, the only significant meQTLs at this locus were identified in AC samples (CpGs6-7), and whilst AEI for COLGALT2 was present in both FC and AC samples, the differential expression was greater in AC (mean ratio = 1.95) than in FC (mean ratio = 1.17). We therefore conclude that the biological mechanism underlying OA risk at this locus is established during development, however, the functional impact at this locus is clearly acquired in later life. Notably, the effect size of mQTLs alone does not appear to be a strong predictor of biological function at this locus.
Conversely, Locus 7 appears to be functionally active during fetal development. Here, 3/6 CpGs cluster within the RWDD2B promoter and are hypomethylated both in development and older age, consistent with the open chromatin state observed across all tissues. Significant meQTLs were observed in FC samples, indicating that the observed DNAm changes are driving a decrease in RWDD2B expression from the beginning of life. RWDD2B encodes the RWD domain–containing protein 2B, about which very little is currently known (25). Proteins containing RWD domains are capable of binding to enzymes including ubiquitin ligases (42). This lack of knowledge regarding the biological function of RWDD2B makes it a worthy target of future functional studies in the context of musculoskeletal disease.
GDF5 (Locus 6) and its regulation have been extensively studied in the context of musculoskeletal development and OA (28,43–47). Identified as a risk gene for OA by the association SNVs rs143383 and rs143384 (48) within the GDF5 5'UTR, early studies into the mechanism behind the regulation revealed AEI for the gene in human aged cartilage and cell models, with the OA risk allele, T, correlating with lower levels of gene expression (28,43,46). More recent, in-depth studies into this locus have utilized murine models, and in vitro reporter assays to identify a Gdf5 enhancer, GROW1, which contains a common derived SNV (rs4911178, G > A) at an otherwise highly conserved position (44). However, to date, the molecular basis of GDF5 expression has not been investigated in primary human fetal chondrocytes. Surprisingly, we did not identify significant AEI for GDF5 in FL or FC samples, however, we did observe a significant imbalance in AC samples, consistent with previous studies (28,43). Therefore, we postulate that, in humans, the differential expression of GDF5 becomes more pronounced throughout the life course, rendering Locus 6 one at which the functional risk of OA is conferred in ageing, with the decreased levels of GDF5 hindering the ability of chondrocytes to maintain and repair the cartilage tissue in older age (49).
Whilst decades of research have been dedicated to understanding the genetic etiology of OA, the clinical exploitation of OA genetic discoveries is still out of reach (50). In this report, we undertook a detailed molecular genetic analysis of well-characterized OA risk loci, using primary human musculoskeletal fetal tissues for the first time. Our data indicate that the functional timepoints of OA risk loci can differ, raising the question of whether causal variants could be classified by whether they confer a developmental or age-acquired risk. The translation of genetic discoveries in the field requires a deep understanding of the molecular mechanisms by which risk-conferring alleles impact their target genes, and the appropriate timepoint for therapeutic intervention, prior to macroscopic structural damage occurring in the joint (11). This is the first study to identify the presence of mQTLs in human fetal cartilage and limb tissues, and our report demonstrates that the functional genetic risk of OA can be laid down during human skeletogenesis. Strides are being made within the field, with the first recent reports of polygenic risk scores (PRS) for the disease (51,52), yet the clinical utility of such systems is still lacking (53). We would encourage the integration of epigenetic data at the loci, along with clinical and biochemical parameters to further advance these tools for patient benefit.
{kind=link}
{kind=link}