Parasite culture and cell lysate preparation
Human foreskin fibroblasts (HFF, ATCC® SCRC-1041™) cells were cultured in DMEM medium containing 10% fetal bovine serum, 100 µg/ml streptomycin and 100 U/ml penicillin. After the cells reached 85% confluency, the virulent WH3 strain tachizoites of T. gondii (type Chinese 1), which dominates in animals and human in China [18], were added to the plates and cultured in CO2 incubator at 37℃. The parasites were purified by three times of washing and centrifugation. Cell lysates were prepared by combining freeze/thaw treatments and ultrasonication followed by centrifugation. The supernatants were collected and stored at -80 °C as WH3 strain parasite antigen (WH3Ag). The cysts of low virulent WH6 strain (type Chinese 1) were harvested from the brain tissues of mice under euthanasia after 6 weeks of cyst gavage.
Animal infection and human serum collection
The 8- to 10-week-old male BALB/c mice (SPF) were purchased from the Animal Center of Anhui Medical University (AMU) and had free access to sterilized water and food under standard conditions. The experiment protocols were conducted with approval of the Animal Ethics Committee of Anhui Medical University ( Permission No. AMU26-081108). The mice were treated strictly in compliance with the Chinese National Institute of Health Guide for the Care and Use of Laboratory Animals. All of the animal experimental procedures were performed in licenced Biosafty II Laboratory. Thirty-five mice were inoculated intraperitoneally with 100 tachyzoites suspended in 100 μL of PBS, and five mice were injected with 100 μL PBS were taken as control. Sera were collected from the animals under euthanasia on days 1, 2, 3, 4, 5, 6 and 7 after infection. Additionally, for infection of low virulent WH6 strain of Toxoplasma, 90 mice were divided into three groups (n=30/group), and were infected with 10, 30, and 60 cysts for each through gavage, taken as the low, medium, and heavy dose infections, respectively. Five animals in each group were sacrificed under anesthesia on days 1, 3, 5, 7, and 14 post-infection. The sera were collected and stored at -80 °C for next use.
Fifty human serum samples were collected from the surplus sera after routine prenatal examination of pregnant women and the children with suspected congenital toxoplasmosis. The sera collection was approved by the Clinical Trial Ethics Committee of the Second Hospital of Hefei, Anhui (Approval No. 2020-Ke-054). The informed consents were obtained from all of the participants or their guardians in the study. Sera were subjected to PCR amplification for Toxoplasma DNAs (Toxoplasma PCR) and the results were taken as golden standards in comparison with Toxoplasma circulating antigen detected by ELISA (Toxoplasma CAg-ELISA) [19] and DELAA test kit.
Preparation of r-SAG1 and its biotin-labeling in vitro
The truncated SAG1 encoding gene (sag1) (forward primer: 5'- GGATCCTTCACTCTCAAGTGCCCTA- 3' and reverse primer: 5'- AAGCTTCCTGCA GCCGGAAACT-3') was amplified using Toxoplasma cDNA as template, ligated to the vector (pET32a), and transformed into E.coli BL21 host cells. The r-SAG1 expression in the host cells was induced by IPTG and the cells were sonicated at 5s intervals and centrifuged at 12 000 rpm for 20 min to obtain the supernatants containing r-SAG1 protein. The r-SAG1 protein with 6×His tags was purified using nickel column (Millipore, USA) from the supernatants. The r-SAG1 protein was separated by 10% separation gel in SDS-PAGE and transferred onto nitrocellulose membrane (NCM), followed by blocking with 5% of milk on a shaker for 90 min. The NCM was incubated with primary antibodies against r-SAG1 (1:1 000 dilution) at 4 °C for overnight and washed with TBS-T for three times followed by incubation with the HRP-conjugated secondary antibody, with slight shaking for 90 min. Sample stripes were visualized using enhanced chemiluminescence. The experimental data were analyzed using Image J 1.46 software.
For biotinylation of the r-SAG1 protein, 1.875 μL 20mM EZ-Link NHS-PEG4-Biotin (Thermo, No. 21329, USA) was added to 0.1 mL 0.5mg/mL r-SAG1 protein at 4 ℃ for overnight, ensuring that one r-SAG1 protein could be labeled with 3-5 biotin molecules. The r-SAG1 protein was dialyzed in PBS to remove excessive NHS-PEG4-Biotin, and NHS-PEG4-Biotin-labeled (biotinylated) r-SAG1 was used for subsequent aptamers screening.
Screening of ssDNA aptamers against r-SAG1 in vitro
Aptamer microsphere library (containing 109 microspheres and 104-105 repeats) was purchased from AM-Biotech, USA. The oligonucleotides of the library differ in 3-D conformation from those commonly used, due to the modifications with Indole-dU (W), Phenol-dU (Y), and Amine-dU(X) to improve the probability of aptamer harvests with high affinity.
The aptamer microsphere library was mixed with 2 mL buffer A (containing 0.5nM BSA and 1nM MgCl2) and heated at 95℃ for 5 min, then cooled at room temperature to create folded ssDNA. M-280 streptavidin-magnetic particles (SMps) (Thermo, No.11205D, USA) were added to the aptamer microsphere library and slightly shaked at 150 rpm at 37 ℃ for 30 min, in order to remove the aptamers that could bind to M-280 SMps. Next, the biotinylated r-SAG1 protein was combined with unused M-280 SMps and then added to the ssDNA library for positive screening of specific aptamers. The M-280 SMps-biotin labeled r-SAG1-aptamer complex was mixed with 50 μL 1nM NaOH and incubated at 65 ℃ for 30 min. Then 40μL 2mM Tris-Cl was added to the complex for dissociation of specific aptamers for secondary screening. The dissociated solution was divided into 3 tubes, with 15 μL each (#1: initial solution control, with additional 135 μL buffer A ; #2: 100nM protein, with additional 30 μL r-SAG1 and 100μL buffer A; #3: positive SMps control, with additional 130 μL buffer A), and was incubated at room temperature for 1h. Five microliters of SMps were added to groups #2 and #3, respectively, and incubated at room temperature for 30min. The groups #2 and #3 were placed on magnetic racks to collect the filtered aptamers. Afterwards, the products of the three tubes (#1, #2 and #3) were used for PCR amplification. The three groups were amplified separately by PCR containing 10 μL 10×PCR buffer, 2.5 mM MgCl2, 0.4 M, with same forward primer (K-FP: 5’-CAGGGGACGCACCAAGG-3’), 0.4 M different reverse primers (K-RP- #1: 5’-ATCACGCAGCACGCGGGTCATGG-3’; K-RP- #2: 5’-CGATG TCAGCACGCGGGTCATGG-3’; K-RP- #3: 5’-TTAGGCCAGCACGCG GGTCATGG-3’), 0.2 mM dNTP, 1U Taq polymerase (Takara, Japan). The PCR was performed using the following conditions: 94 ℃for 60s, followed by 94℃for 30s, 65 ℃for 30s, and a final extension of 60s at 72 ℃. The PCR cycle numbers of the three groups were set as 10, 15, 20, and 25, respectively and the cycle number was taken when all of the three groups presented positive PCR bands. The PCR products were separated by 5% agarose gel electrophoresis. The aptamers in the PCR-generated products were sequenced, and modified with Indole-dU (W), Phenol-dU (Y), and Amine-dU(X) after site positioning using AM Cloud Intelligent Software(AM Biotech. Co. Ltd., USA). During synthesis, the four aptamers were simultaneously biotinylated.
Optimization of WH3Ag concentration binding to aptamers
A concentration of 1.56 μg/mL of bovine serum albumin (BSA) and 1.56 μg/mL of recombinant dense granule protein 15 of T.gondii (rGRA15, previously prepared in laboratory) [20] were used as the negative controls for the determination of affinity of aptamers to n-SAG1 protein of Toxoplasma. The WH3 strain parasites were frozen and thawed 5 times in liquid nitrogen and sonicated at 3s intervals at 20W. The sonictated lysates were centrifuged at 5000rpm for 20min and the supernatants served as WH3Ag. The WH3Ag was serially diluted to 0.78, 1.56, 3.125, 6.25 , 12.5, 25, 50, 100, and 200 μg/mL in 0.1M Na2CO3 and NaHCO3 (pH9.6). After washing with 0.01 M PBS-T for five times, blocking buffer (0.5% BSA-PBS, pH 7.2) was added to each well and incubated at 37 ℃ for 2h. The plate was washed three times with PBS-T (pH 7.4), 100 nM biotinylated aptamers were added to each well and incubated at 37 ℃ for 1h. Then, the plates were washed three times with PBS-T and incubated with 100 μL of streptavidin diluted to 1:10 000,1:12000,and 1:15000, which had been conjugated to HRP, at 37℃for 1h. The plates were washed with PBS-T five times and tetramethylbenzidine(TMB)was added and allowed to stand for 15 min. The color development was terminated using stop solution (H2SO4) and the absorbance at 450 nm was measured. The optimal concentration of WH3Ag was defined as 1.56 μg/ mL.
Binding affinity of the aptamers to n-SAG1 in WH3Ag
To detect aptamers affinity to the n-SAG1, a 96-well plate was coated with 1.56 μg/mL WH3Ag and incubated at 4 ℃ for overnight. The four aptamers were diluted at 0.00, 1.56, 3.125, 6.25, 12.5, 25, 50, 100, and 200 nM, respectively. After washing, 100 μL HRP-streptavidin (in dilution of 1:10 000, Shenggong Biol Co, Ltd., China) was added to each well and incubated at 37 ℃ for 1h, the plate was washed five times with PBS-T, and TMB was added. The plate was allowed to stand for 15min. The affinity of the four aptamers to WH3Ag was analyzed. The examinations of all samples were performed in triplicate and the OD values were measured at a wavelength of 450 nm (OD450).
Optimization of test performance and determination of cut-off value
A 96-well plate was coated with a mouse serum of acute Toxoplasma infection which were serially diluted from 1:5 to 1:80 for optimization of test dilution. A negative control was set up in parallel. After incubation for 2h at 37 ℃, the plate was incubated at 4 ℃ for 12h. After 5 times of washing, 1.56 nM aptamer-2 was added to each well and incubated at 37 ℃ for 1h. After washing, the HRP-labeled streptavidin with a dilution of 1:10 000 was added and incubated at 37 ℃ for 1h. After washing as before, TMB was added after washing and allowed to stand in the dark for 15min, and then 50μL stop solution was added to each well. The specimens of all experimental groups were repeated three times and the OD values were measured at a wavelength of 450 nm. The dilution at the concentration of 1:10 presented the highest ratio of P/N(positive/negative). The optimal dilution in 1:10 for mouse sera was used and the OD450 value of 20 serum samples of normal mice was measured. The cut-off value was determined by mean±2SD and was used in the subsequent tests of mouse sera under optimized experimental conditions.
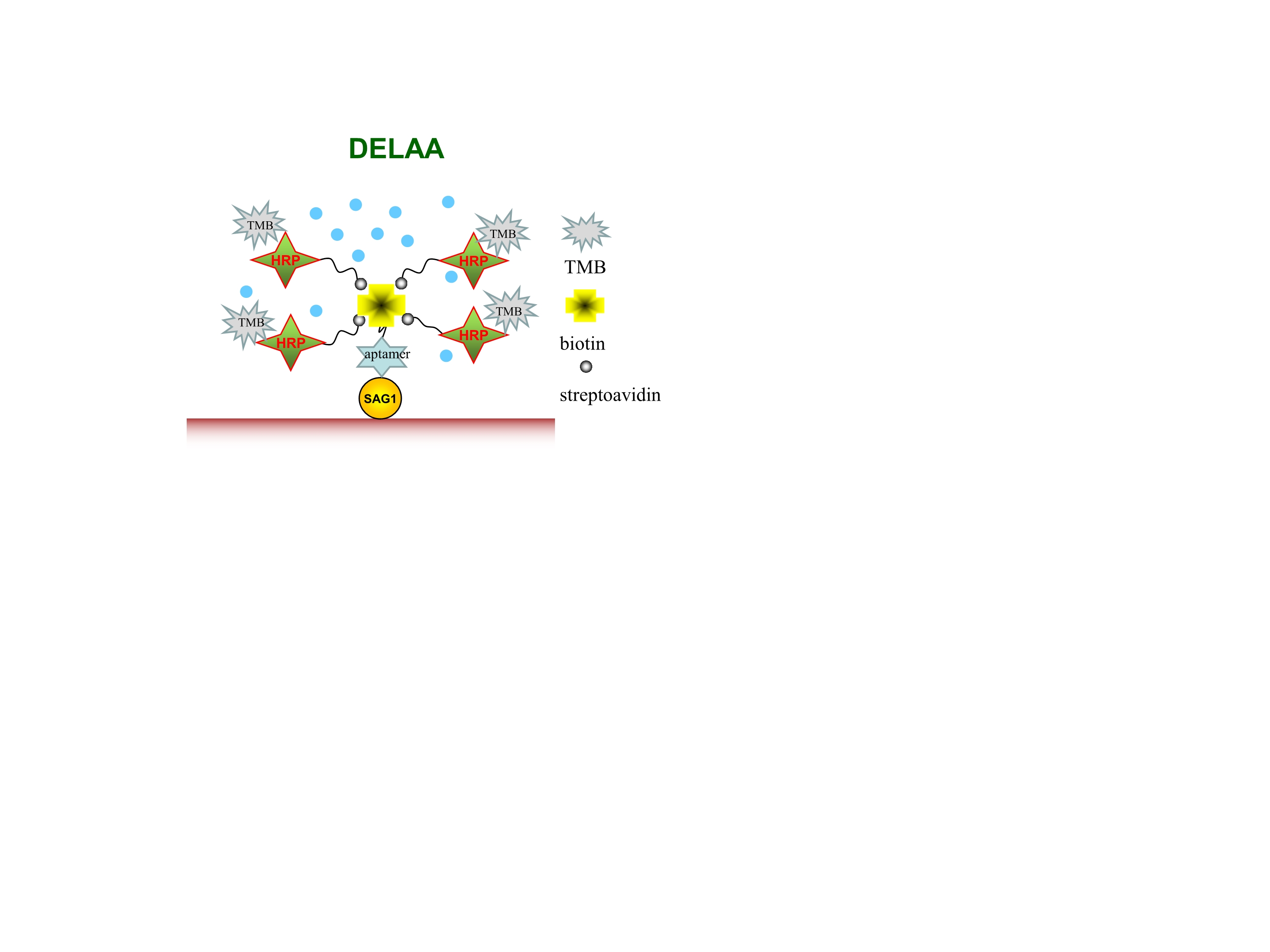
Direct enzyme-linked aptamer assay (DELAA) for human serum detection
Twenty seronegative human sera ( neither IgG nor IgM against T.gondii examined by the commercial ELISA kit (NA Multi-Lyte ToRCH,Zeus Biotech Co. Ltd, China) from the hospital were tested to calculate the threshold value by DELAA. Serum samples of 50 cases of acute Toxoplasma infection (with positive IgM but negative IgG against T.gondii) were collected based on screening of suspected individuals with the ELISA kit. Among them, fifteen positive and 35 negative samples that had been confirmed by PCR for detection of Toxoplasma DNAs (Kanglang Biotech Co. Ltd, China)were re-examined with the commercial kit of Toxoplasma CAg-ELISA for detection of Toxoplasma circulating antigens (Combined Company, Shenzhen, China) [21] and DELAA, respectively, to detect circulating antigens or n-SAG1 of T.gondii. The serum samples were examined in triplicate and three tests were performed per sample.
Statistical analysis
Data were subject to normal distribution and statistically described as mean ±standard deviation, and differences between groups were compared using one-way ANOVA. The chi-square test was used to compare the sensitivity and specificity of Toxoplasma CAg-ELISA and DELAA (p<0.05 or p<0.01 indicating statistical significance). All analyses and graphing were performed using GraphPad-Prism software.
{kind=link}