1. Trem2 is primarily activated in repopulated NAM during NASH progression
AMLN, a NASH diet containing 40% trans fat, 20% kcal fructose, and 2% w/w cholesterol, induces rapid and robust weight gain in wild-type or Trem2Flox mice10.The 30 weeks fed liver exhibits prominent features of NASH, including lipid accumulation, chronic inflammation, and portal and perisinusoidal fibrosis (1A). The corresponding choline-deficient L-amino acid-defined CDAHFD60 high-fat diet model is a modified MCD model that can induce severe hepatic steatosis and fibrosis in a short period of time without causing weight loss in mice (1A). We used the two feeding models described above to investigate the role of myeloid Trem2 in the progression of NAFLD and NASH. Xiong et al. have previously attempted to uncover the identity of NASH-associated Trem210. To further explore the immune cell subsets for NASH-associated Trem2 localization, we performed cyTOF on CDAHFD60-induced fibrotic livers. Trem2 was found to be significantly enriched in MHCII+CD9+CD11c+CX3CR1+OPN+NASH-associated macrophages which is upregulated in CDAHFD60-NASH livers, similar to previous descriptions(1B-D)7,10. In addition, Trem2 transcript levels were significantly up-regulated in AMLN- and CDAHFD-fed NASH livers, along with higher protein abundance(1E-G). Immunofluorescence shows that trem2 is expressed in both CD11b+TIM4+ repopulating macrophages and CD11b+TIM4− monocyte-macrophages, and correlated with αSMA expression in CDAHFD60-fed NASH livers (1H).
2.Myeloid Trem2 drives hepatic macrophage niche remodeling during NASH progression
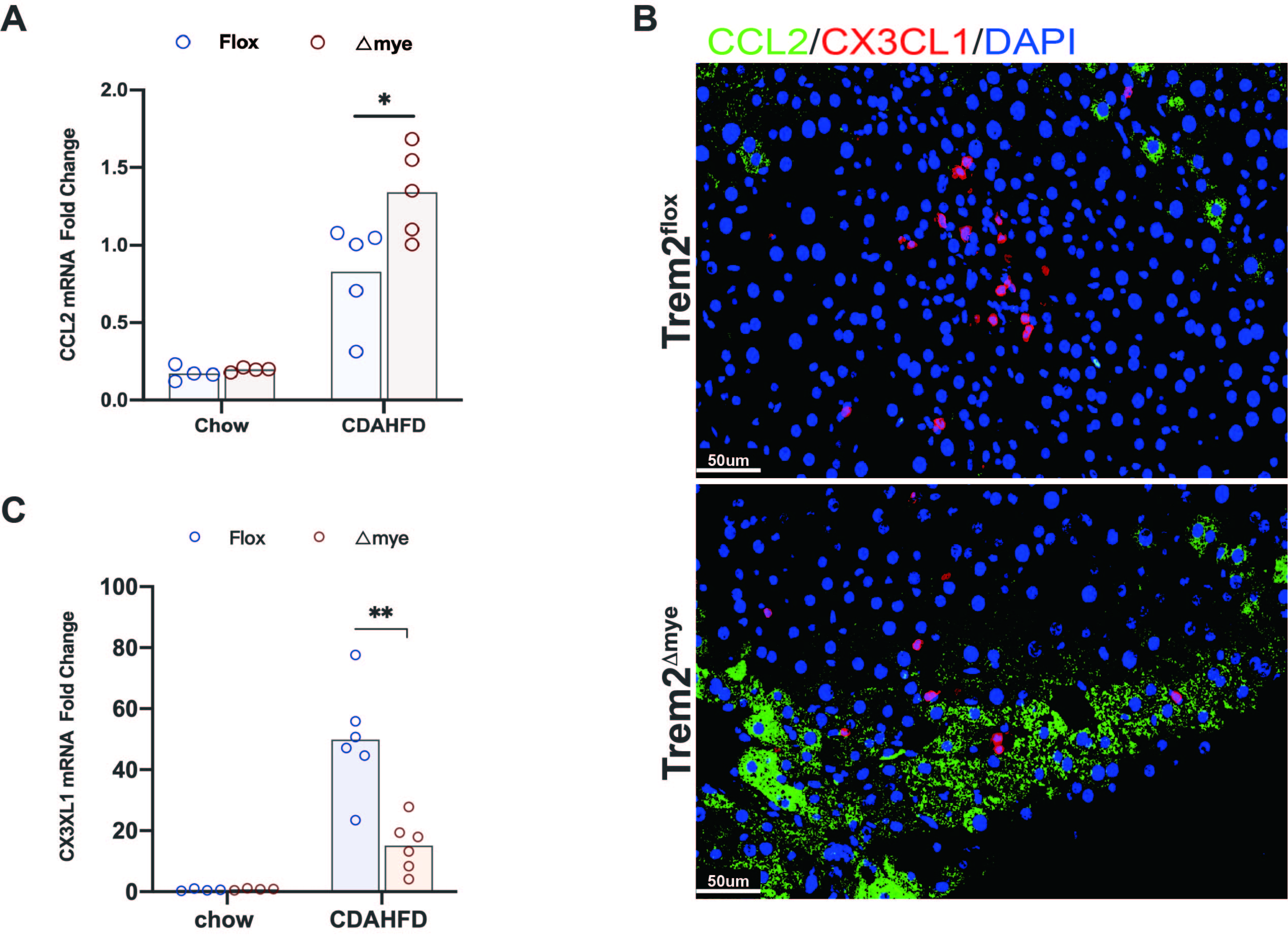
NASH is accompanied by dynamic changes in the macrophage pool, characterized by exhaustion of kupffer cells and backfill of monocyte-derived macrophages10. The changes of different phenotypes and phase-defined cluster in CDAHFD60 NASH livers deficient or not deficient of myeloid Trem2 were analyzed by cyTOF assays. Indeed, the accumulation of recruited CCR2+Ly6Chi iMFs in response to NASH activation were further exacerbated by depletion of myeloid Trem2 (2A), the flow landscape of monocyte-macrophage differentiation was revealed by t-SNE(2B). Contrastive analysis revealed a significant accumulation of infiltrating macrophages of Ly6ChiCX3CR1lo defined by Cluster22 in Trem2△mye NASH livers(2B,C,H) and was characterized by upregulated Ki67 abundance (2B,D). In fact, the heatmap shows that Cluster20/21/22/24, defined by the expression levels of CD11b, Ly6C, CX3CR1, Tim4, F4/80, are activated by NASH and represent different reprogramming stages of macrophage subclusters(2E), Infiltration abundance and proportion were quantified (2F). An additional group of Ki67+Ly6ChiF4/80− monocytes appeared in the NASH liver of Trem2△mye(2B), which was assigned as Cluster20(2G).Correspondingly, both the frequency and number of Ly6ChiCX3CR1hi transitional-macrophages and Ly6CloCX3CR1hi repairing-macrophages were downregulated in NASH livers in response to absence of myeloid Trem2 (2B, I, J). CCL2 and CX3CL1 are chemokines indicative of monocyte recruitment and reprogramming of pro-resolution macrophages. The transcript level and protein expression of CCL2 in NASH livers induced by CDAHFD60 feeding for 8 weeks were significantly up-regulated in the absence of myeloid Trem2 (S1A,B). The expression of CX3CL1 was corresponding suppressed(S1B,C).The above results suggest that myeloid Trem2 has a nonnegligible contribution to drive the reprogramming of NASH-recruited macrophages.
3. Absence of myeloid trem2 exacerbates AMLN-hepatic steatosis
Oil red staining of livers fed AMLN for 16 and 30 weeks showed increased lipid accumulation in the livers of myeloid Trem2-deficient mice. In contrast, Rosa26 conditionally overexpressed Trem2 mice had lower lipid accumulation by 30 weeks.(3A). We then performed a comprehensive lipidomics analysis of AMLN-fed livers. PCA principal component analysis and OPLS-DA analysis determined that the lipid metabolism profile of the NAFLD liver of Trem2△mye mice was significantly different from that of Trem2Flox (3B-C), in which Trem2△mye led to significant upregulation of 385 lipid metabolites in AMLN liver, and 46 significant down (3D), and the significantly up-regulated metabolites included triglycerides TG, CE(Consistent with previous research4) and ceramides(3E-G, 3I). However, liver weight did not appear to have a statistically significant change at 16 weeks of AMLN (3H). In vitro experiments in the presence or absence of Trem2 BMDM and AML12 hepatocyte line or primary hepatocyte co-culture system add palmitic acid to induce AML12 steatosis. Nile red staining and oil red staining showed that Trem2 mKO causes hepatocytes to accumulate more triglycerides (3K, 3L).
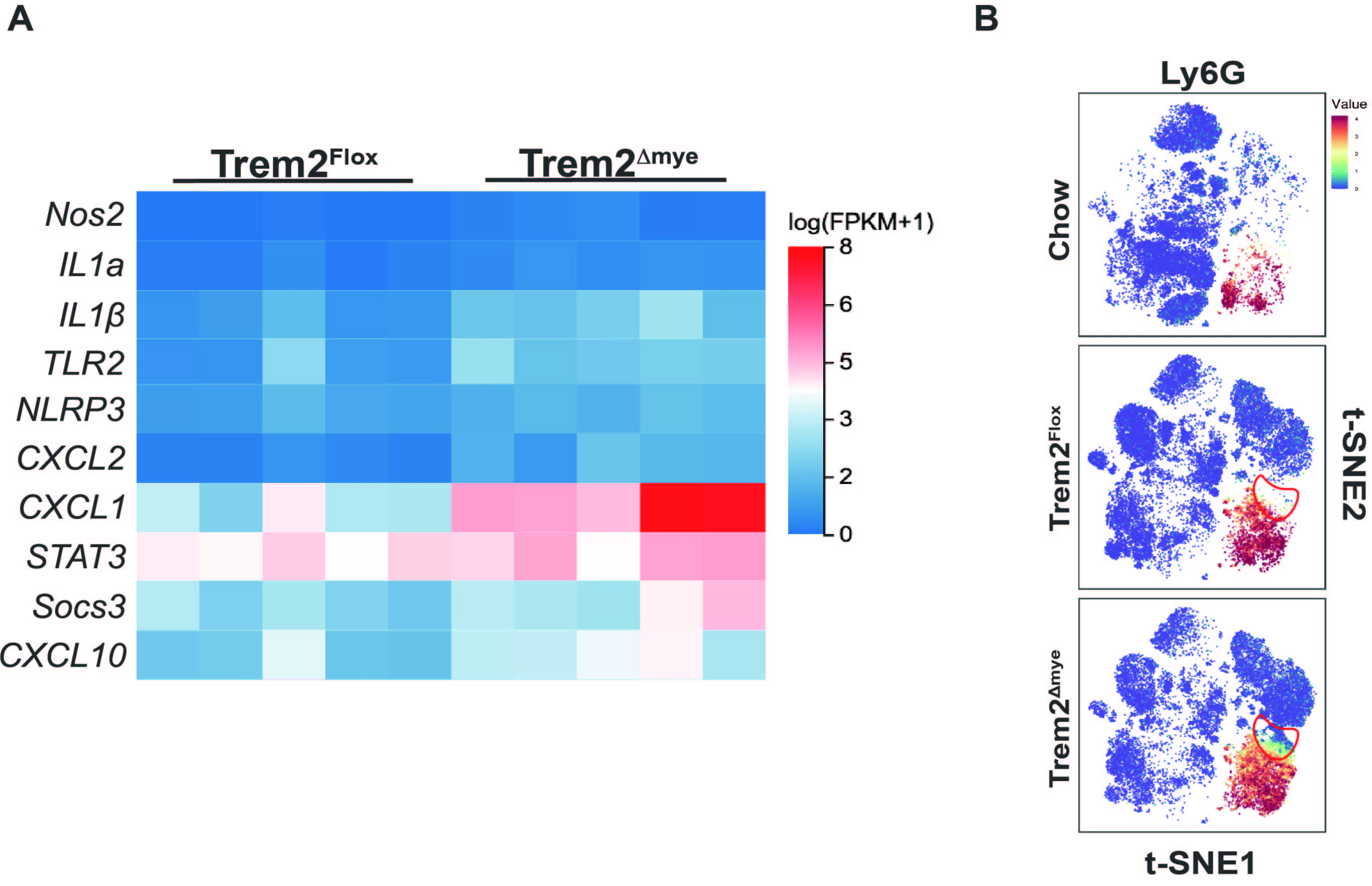
We speculate that knockout of myeloid trem2 exacerbates hepatic steatosis due to increased inflammation-induced. In other words, NAFLD-related inflammation would be exacerbated by the termination of trem2-dependent reprogramming instructions in response to AMLN. As expected,Trem2△mye increased the transcription and secretion of pro-inflammatory mediators such as IL1β,NLRP3 and Socs3 in NAFLD livers fed AMLN for 16 weeks(S2A).Notably, TLR2 expression was exacerbated by Trem2△mye. Not only that, neutrophil recruitment of CXCL1/2 was upregulated by myeloid Trem2 deficiency(S2A). t-SNE showed that Trem2△mye indeed increased iNOS + neutrophil infiltration in NAFLD(2B,S2B).
S3: Knockout of myeloid Trem2 suppresses high-fat feeding-driven systemic obesity in mice
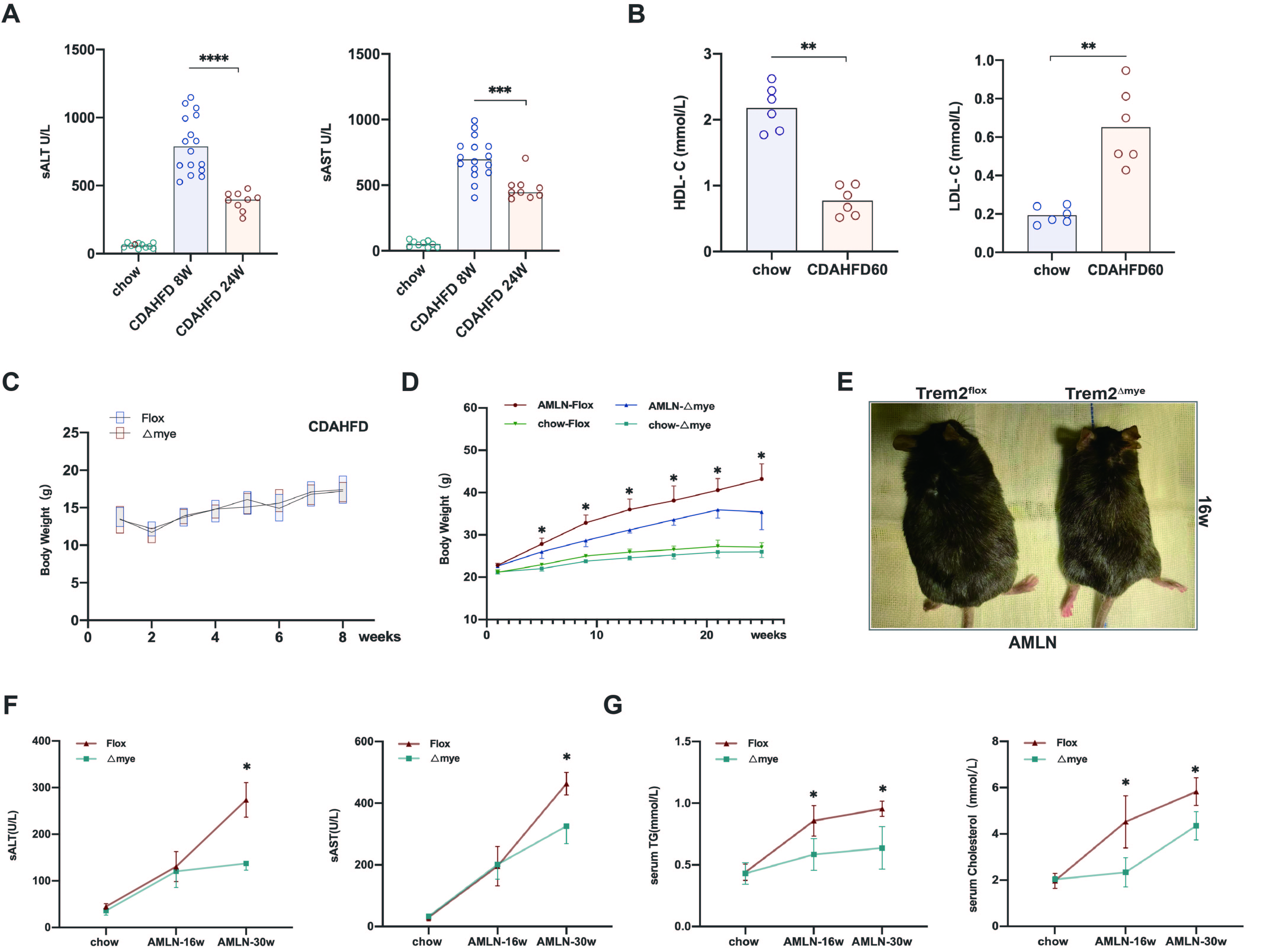
CDAHFD60-fed mice for 8 weeks developed significant hepatic inflammation(S3A), accompanied by down-regulation of serum HDL-C and up-regulation of LDL-C (S3B), but neither Trem2Flox or Trem2△mye mice fed with CDAHFD60 cause significant weight change within 8 weeks(S3C). Notably, although a previous study showed that global gene ablation of Trem2 in mice resulted in systemic hypercholesterolemia, body fat accumulation, and glucose intolerance 8,myeloid-specific Trem2-deficient mice were strongly resistant to AMLN-feeding induced rapid weight gain and body obesity trends (S3D, E). Although the secretion of inflammatory factors was exacerbated in the liver of myeloid Trem2-deficient mice fed AMLN for 16 weeks, serum ALT and AST levels were not significantly different from those of Flox mice (S3F). When AMLN progressed to 30 weeks, Trem2△mye mice had significantly reduced serum ALT and AST compared to Flox, reflecting that myeloid Trem2 deficiency somehow protected hepatocytes from damage of inflammation and lipid induction. Not only that, NAFLD-NASH Trem2△mye mice also had lower serum triglyceride levels and cholesterol levels (S3G). Overall, although the absence of Trem2 slightly increased NAFLD-induced hepatic inflammatory factor secretion, systemic metabolic disturbances as reflected by body weight, serum triglycerides, and serum cholesterol were alleviated.
S4. Myeloid Trem2 regulates hepatic fatty acid synthesis and degradation
To explore the mechanism by which myeloid Trem2 is involved in regulating hepatic steatosis, we performed whole-transcriptome RNA-sequencing of the livers of Trem2Flox or Trem2△mye mice at 16 weeks of AMLN. KEGG analysis based on RNAseq results assigned PPAR signaling pathways and fatty acid degradation, among others, as enriched gene sets for which knockdown of myeloid Trem2 resulted in significant alterations(S4A), similar to the enrichment results by comprehensive lipid analysis(S4B). Trem2△mye decreased the expression of lipid synthesis-related genes including PPARγ and Sorbs1 in livers upregulated by AMLN feeding (S4C). Plin4, a target gene of PPARγ, can promote lipid droplet formation and inhibit fatty acid degradation, also downregulated by Trem2△mye(S4C). On the other hand, Trem2△mye also significantly inhibited the expression of fatty acid β-oxidation pathway related genes CPT1a, Acaab1, Ehhadh, cyp4a10/14(S4E,F).Aldh3a2 and Cyp4a12 are directly and actively regulated by Pparα, responsible for the detoxification of toxic lipids (S4C-D13 14). Although either RNA-sequencing or qPCR characterization of AMLN liver showed that Trem2△mye did not cause significant changes in Pparα expression, the expression of Aldh3a2 and Cyp4a12 was significantly down-regulated in mice. Importantly, liver carnitine levels were also significantly downregulated(S4G,H). Carnitine is a carrier for transporting long-chain fatty acids, which can promote the oxidative decomposition of fatty acids.
4. Trem2 △mye is more likely to induce liver fibrosis in NAFLD mice, but plays a bidirectional role in the ongoing progression of liver fibrosis
Trem2hi macrophages in NASH livers based on the CDAHFD model have previously been shown to be involved in the regulation of high expression of genes associated with extracellular matrix remodeling 10. Therefore, we hypothesized that myeloid Trem2 may be responsible for driving the activation of HSCs to promote the progression of liver fibrosis. Sure enough, in our Trem2△mye livers fed the CDAHFD diet for 8 weeks, the fibrosis-inducing genes αSMA, Cola1/2 were significantly down-regulated compared with Trem2Flox livers (4A),but no statistically significant differences in the extent of liver fibrosis observed by Sirius Red and Masson staining was puzzled us (4B-D), so we further extended our study; concomitant increased inflammation in the liver of Trem2Δmye AMLN-NAFLD, The transcription of Acta2, Col1a1/2 was slightly up-regulated relative to Flox liver, while Gata4 and Pparγ, which promote fibrosis regression and dormancy, were relatively down-regulated (4E). Consistent with the results of RNA-Seq, histological analysis showed that AMLN-NAFLD livers lacking myeloid Trem2 were relatively more likely to develop significant fibrosis (16weeks—Flox/△mye:1/5:3/5) (4F), this may be due to the exacerbated accumulation of pro-inflammatory cells associated with Trem2△mye hepatocyte steatosis increasing the susceptibility to fibrosis, as hepatic fibrotic cords at 16 weeks of AMLN are enriched around lipid vacuoles. However, when AMLN was fed to 30w, the induced levels of Col3a1, Col1a1/2, which mediate extracellular matrix remodeling, were not significantly different between the two groups and were even down-regulated in Trem2△mye liver (4G);
Based on this conflicting background, we induced myeloid Trem2 overexpression (4H) by injecting Trem2creERTRosa26Tdtomato with Tamoxifen to investigate how myeloid Trem2 manipulates NASH-associated fibrosis outcomes in relatively independent stages of hepatic fibrosis after 16 weeks AMLN feeding of stable hepatic steatosis. Tamoxifen-induced overexpression efficiency was determined by flow cytometry of the tdTomato fluorescence intensity of isolated macrophages (4I). Addition of tamoxifen after 16 weeks-AMLN to induce myeloid trem2 overexpression resulted in a significant increase in liver fibrosis at the experimental endpoint of 30 weeks-AMLN(4J,K), suggesting that precluding intervention on the chronic inflammation that accompanies steatosis, myeloid Trem2 promotes the induction of fibrosis through additional mechanisms. In conclusion, although Trem2△mye NAFLD is more likely to progress to NASH, myeloid Trem2 appears to have a bidirectional regulatory effect on the continued progression of fibrosis.
S5. OPN expression and hCLS formation in NASH-associated macrophages are associated with myeloid Trem2
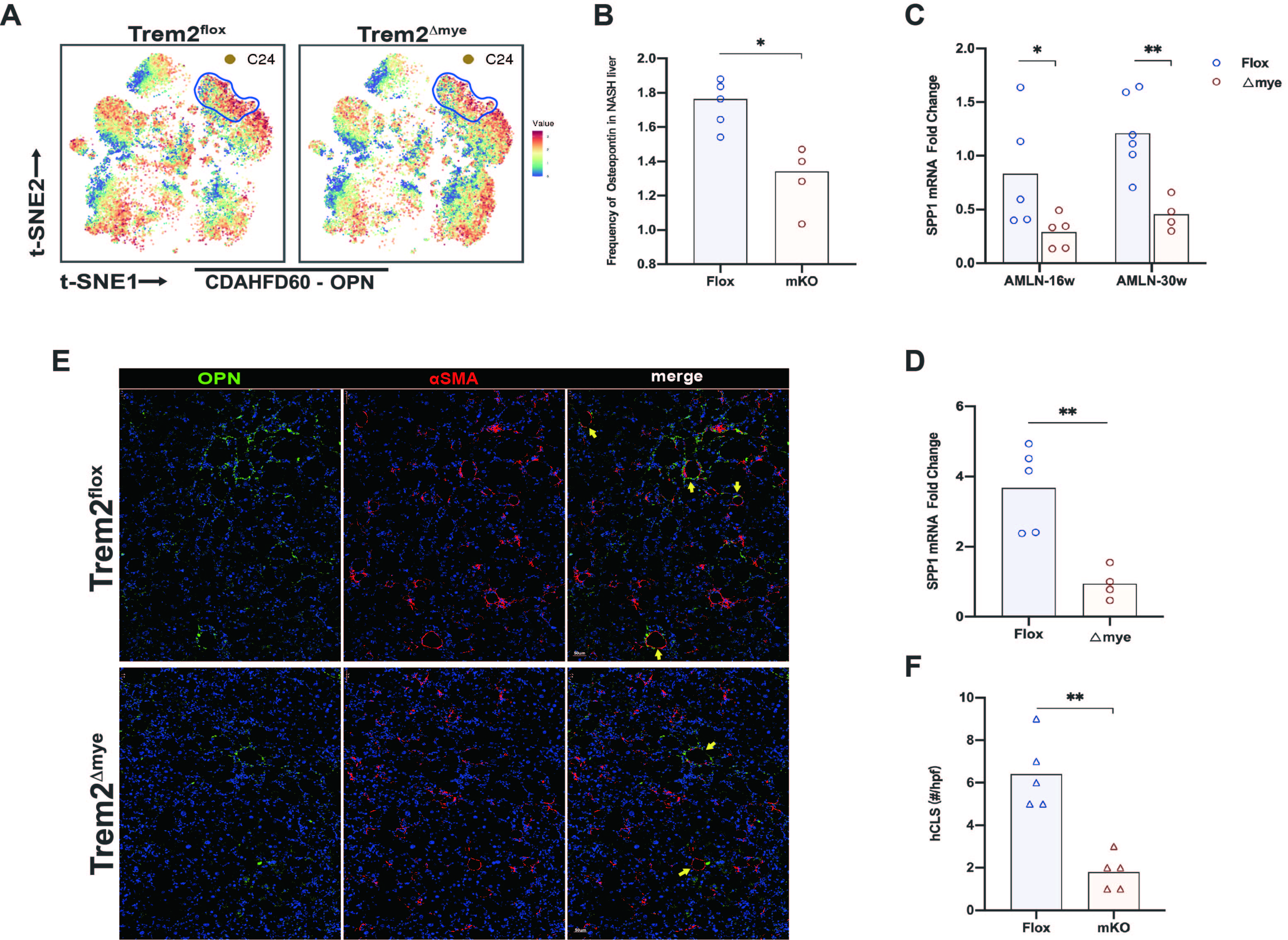
NAM labeled by osteopontin is responsible for reflecting the progression of liver fibrosis in NASH 15. Currently, OPN is mainly believed to promote liver fibrosis16. Cytof identified that knockout of myeloid Trem2 significantly reduced the abundance of osteopontin (S5A,B). The transcript level of SPP1, which encodes OPN, was also relatively significantly downregulated in Trem2△mye NASH livers (S5C,D).
In addition, Trem2△mye did significantly reduce the formation of hCLS in NASH liver while alleviating liver fibrosis (S5E,F). hCLS (hepatic coronary structure) is a ring-shaped structure formed around lipid bullae during NASH liver tissue remodeling and fibrosis induction. Liver fibrosis is accompanied by increased hCLS formation (S5E). Obeticholic acid (OCA), an FXR agonist, has been shown to improve fibrosis in NASH, significantly reducing hCLS formation, suggesting a role for hCLS in driving fibrosis 17. Overall, more studies may be needed to elucidate whether frequency of OPN and formation of hCLS acts as a facilitator for NASH-related liver fibrosis progression.
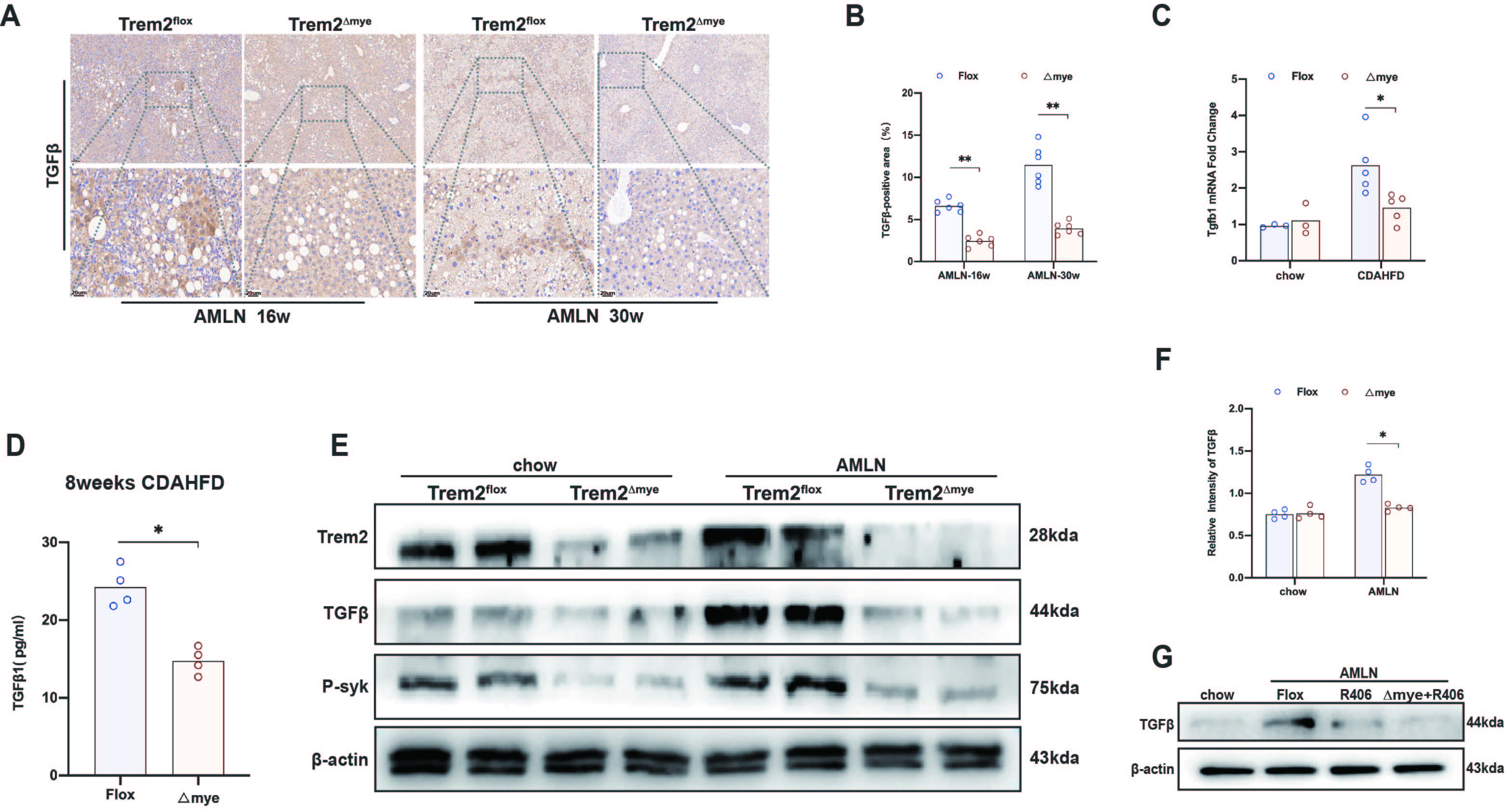
5.Myeloid Trem2 activates TGFβ in a P-syk-mediated efferocytosis program to induce HSC activation
TGFβ is significantly activated in AMLN-NAFLD/NASH liver(S6A). When we blocked myeloid Trem2, the expression of TGFβ was significantly reduced in AMLN/CDAHFD-NASH livers (S6A-F). TGFβ1 is not only the main product of the efferocytosis program18, but also the most potent activator of HSCs 19. Therefore, we speculate that the inhibition of TGFβ activation by Trem2△mye is mainly due to the impairment of the efferocytosis program.
Knockdown of Trem2 has been shown to inhibit Mφ efferocytosis in multiple inflammatory models. But its role as "find me" or "eat me" remains unknown. We stimulated primary HSCs in vitro with efferocytosis culture supernatants of BMDM in the presence or absence of Trem2. Deletion of BMDM Trem2 significantly inhibited efferocytosis-induced activation of mouse primary HSCs(5A). NASH livers from myeloid Trem2-deficient mice fed AMLN for 30 weeks had more residual TUNEL-labeled apoptotic cells (5B). To demonstrate whether the accumulation of apoptotic cells is due to Trem2△mye impairing efferocytosis, we injected Celltracker Blue dye-labeled dexamethasone -pretreated apoT through the tail vein of mice. We found that the livers of Trem2△mye mice retained more blue dye-labeled apoT that was not phagocytosed by F4/80 + macrophages (5C). Arg1 is necessary for metabolic maintenance of sustained efferocytosis and release of mediators of resolution of inflammation20, and CDAHFD-induced hepatic Arg1 activation is inhibited by Trem2△mye(5D,E). In addition, PGE2 is essential for polarization and efferocytosis of the anti-inflammatory phenotype. Lipidomic analysis revealed that hepatic PGE2 production was also significantly reduced in Trem2△mye AMLN mice (5F).
As we envisioned, Trem2△mye not only reduced TGFβ protein secretion in efferocytosis supernatants in vitro (5M), but also inhibited the expression of TGFβ latency-associated peptide LAP in BMDM undergoing phagocytic program(5G). The expression of TGFβ1 latency-related peptide (LAP) in the immunosuppressive environment constructed in vitro was also reversed by the absence of myeloid Trem2 (5G); also the expression of phosphorylated syk (5J);P-syk is important for macrophages to complete the phagocytic program of efferocytosis21. Additional OxPAPC stimulation exacerbates P-syk activation in efferocytotic macrophages(5K,L). The absence of Trem2 not only reversed the efferocytosis-driven phosphorylation of syk at Tyr525/526, but also caused BMDM to be unresponsive to OxPAPC stimulation(5K,L). The addition of R406 to Trem2△mye did not further reduce the content of TGFβ(5M), indicating that the inhibitory effect of Trem2△mye on the efferocytosis program of TGFβ is dependent on the blockade of the syk phosphorylation pathway.
This seems to be very similar to the regulatory role played by MerTK, which is also a receptor associated with macrophage phagocytosis22. Indeed, BMDM-induced MerTK activation in response to efferocytosis and efferocytosis supernatants was also significantly inhibited by the absence of Trem2 (5H).
Trem2△mye not only reversed the expression of TGFβ, but also reversed the expression of p-syk in AMLN liver(S6E). We next sought to elucidate whether the regulation of TGFβ by myeloid-derived macrophage Trem2 in vivo was dependent on the p-syk. Phosphorylation of macrophage syk is known to be critical for NASH-associated TGFβ activation 22. Similar to Trem2△mye, the syk inhibitor R406 also reduced the levels of TGFβ1 in AMLN liver, but further inhibited the activation of syk on the basis of Trem2△mye had no significant effect on the expression of TGFβ1(S6G).
6.NASH livers lacking myeloid Trem2 are less likely to progress to HCC
Liver fibrosis, an important measure of NASH, predicts a huge potential risk of liver cirrhosis and HCC progression. Not only were there a large number of αSMA+/Col1A1 fibrous scars that co-localized with Trem2 in NASH-progressed tumors, but also the cord-like fiber bundles surrounding the tumor mass were also significantly associated with Trem2 expression(6A, B). After C57 mice were fed CDAHFD for 24 weeks, the appearance of NASH-HCC was considered a significant event with a low degree of variability (CDA24w: 14/15), whereas AMLN mice had a relatively high degree of variability in tumor progression (AMLN 48w: 3/10) (6C, D). Compared with Flox mice, the incidence of NASH-HCC (5/5:3/5) in Trem2△mye mice fed CDAHFD for 24 weeks was significantly reduced (6E), and the tumor burden in Trem2Δmye mice was significantly reduced (6F, G). By examining the adjacent tissue of NASH-HCC in the two groups of mice, it was found that myeloid Trem2-deficient HCC mice had relatively remission of fibrosis progression in the adjacent tissue of HCC mice (6H,I). In addition, remission of HCC progression may Changes in the NASH immune profile resulting from Trem2△mye, in which the abundance of immune cell markers associated with NASH progression, including CXCR2, were significantly downregulated in Trem2△mye liver (6J). Targeting CXCR2 has proven promising in the immunotherapy of NASH-HCC23;
7.Deficiency of myeloid Trem2 reverses expanded ehaustive Eomes + CD8 + T cells and Tregs in NASH liver
Loss of hepatic CD4 + T cells by NAFLD induces HCC progression24. Indeed, the abundance of CD4 + T cells in NASH livers as NAFLD advanced was significantly reduced compared to control chow diet-fed Lean livers, however the frequency changes of CD4+T cells in NASH livers were not affected by Trem2Δmye (7A). Nonetheless, the expression frequency of NASH-increased CD8 + T cells was reversed by Trem2Δmye (7B). Prophylactic depletion of CD8 + T cells alleviates liver injury and HCC incidence in 30% choline deficient high-fat CDAHFD diet mice suggesting that CD8 + T cells drive NASH-HCC progression25. So is the abundance of clusters mainly enriched in CD8 + T cells affected by Trem2△mye? Knockdown of myeloid Trem2 did not appear to have a significant effect on the abundance of Ly6ChiCD11c+CD8+T cells represented by Cluster10, but reversed the frequency of two CD8 + T cell clusters, C13 and C14, which were upregulated in response to NASH, Trem2△mye also reverse Tregs frequency(7C-H,L). Depletion of Tregs inhibits NASH progression to HCC26. Cluster13 is Eomes+Ki67loCD27+CD8+T cells, consistent with previously described CD8+PD1+ HCC-promoting T cells, while Cluster14 is defined as CD103+CD11c−CD27+CD8+T cells. The expression abundance of Eomes and CD103 was also reversed by Trem2△mye (7I-K). We next wanted to test whether the Eomes + CD8 + T cells reversed by Trem2△mye were predominantly CD8 + T cells with a depleted phenotype. PD1 was significantly activated in Eomes + CD8 + T cells upregulated in NASH livers reversed by Trem2△mye, whereas Trem2△mye decreased the number of PD1-expressing CD8 + T cells and the corresponding abundance of PD1(7M). Immuno- fluorescence showed that CD8 + T reversed by Trem2 were basically positive for PD1 (7N).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}