Materials, cells and animals
All siRNA duplexes were synthesized by GenePharm (Shanghai, China). The sequence of human siMMP-9: 5'-CUA UGG UCC UCG CCC UGA ATT-3', antisense: 5'-UUC AGG GGC GAC CAU AGT T-3'; the sequence of rat siMMP-9: 5'-GGG CUU AGA UCA UUC UUC ATT-3', antisense: 5'-UGA AGA AUG AUC UAA GCC CAG-3'; the sequence of negative control siRNA (siRNA): 5'-UUC UCC GAA CGU GUC ACG UTT-3', antisense: 5'-ACG UGA CAC GUU CGG AGA ATT-3'; siRNA labeled with FAM was abbreviated as siFAM; siRNA labeled with Cy5 was abbreviated as siCy5. Human keratinocyte cells (HaCaT cells, Catalog No. GDC106) were purchased from the China Center for Type Culture Collection (Wuhan, China) and cultured in DMEM (Thermo Fisher, MA, USA) containing 10% FBS. All cells were maintained under 5% CO2 at 37 °C (Thermo Fisher, MA, USA). Sprague Dawley (SD) rats (6 weeks, male) were purchased from the Guangdong Medical Laboratory Animal Center (Guangdong, China) and maintained under a specific pathogen-free barrier facility at the Laboratory Animal Center of Sun Yat-sen University. All in vivo procedures were carried out at the Laboratory Animal Center of Sun Yat-sen University. Protocols described in this study had been approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University (No.00187524 and No.00187525).
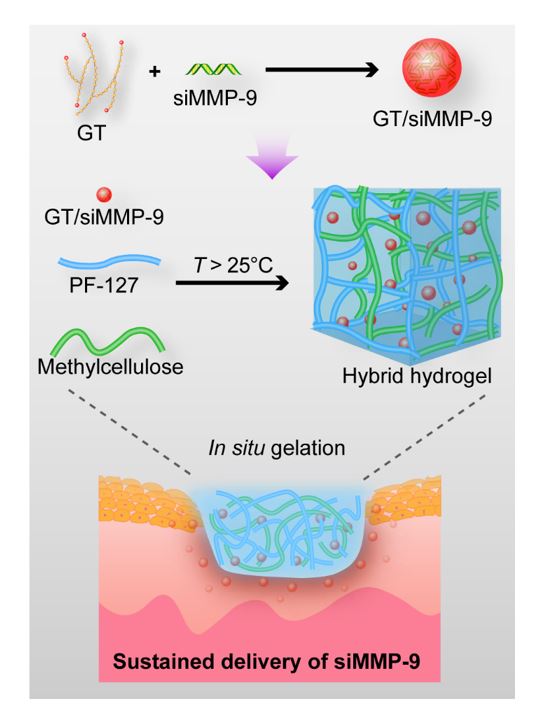
Synthesis and characterization of GT
GT was synthesized by conjugating triethylenetetramine (Aladdin Biochemical, Shanghai, China) to the hydroxyl groups of glycogen (Sigma-Aldrich, Darmstadt, Germany) using the CDI activation method[32]. In brief, glycogen (0.10 g, 0.62 mmol glucose units) was dissolved in 10 mL of anhydrous DMSO, then CDI (5.58 mmol, 0.90 g, Bailingwei Technology, Beijing, China) was added under nitrogen and the mixture was stirred for 1 h. triethylenetetramine (5.44g, 37.2 mmol) was subsequently added and reacted at room temperature for 24 h. The solution was dialyzed against distilled water (molecular weight cut-off ; MWCO:14,000) for 3 days and lyophilized. Fourier transform infrared (FTIR) spectra was recorded on a Nicolet/Nexus 670 FTIR spectrometer (Thermo Fisher, MA, USA) at a resolution of 4 cm-1 by the KBr method. 1H nuclear magnetic resonance (NMR) spectra was recorded on a Varian INOVA500NB NMR Spectrometer (Varian, USA) at 500 MHz, using the signal at δ 4.67 ppm for HDO as an internal standard.
Preparation of the GT/siRNA complex
GT was dissolved in nuclease-free water at 2.15 mg/mL and was then filtered through a Millipore filter (pore size 0.22 μm). Different volumes of GT solutions were mixed with siRNA, the weight ratio of GT to siRNA was varied from 0.5:1 to 30:1. These mixtures were then dispersed in nuclease-free water or opti-MEM (Thermo Fisher, MA, USA) and adjusted to the same final volumes, and the final concentration of siRNA was 0.54 μg/mL. The mixture was incubated for 30 min at room temperature before use. As shown in Table S1, Different siRNA, i.e. siMMP-9, siFAM and siCy5, were used in different experiments. The X in GT/siRNAX represents the weight ratio of GT to siRNA.
Characterization of the GT/siMMP-9 complex
The zeta potential and size of the complex were determined using ZetaPALS (Brookhaven, USA) at 25 °C with a 90° scattering angle by dynamic light scattering (DLS). The complexation of siMMP-9 to GT was also confirmed by agarose gel electrophoresis. GT/siMMP-9 complexes was suspended in nuclease-free water, 3 μL 6× gel loading buffer was added to each sample and 15 μL of the mixture was loaded onto each well in 4% agarose gel with 1× GoldView II nuclear staining dye (Solarbio, Beijing, China). Electrophoresis was run in TBE buffer (pH 8.3) at 80 V for 20 min and the gel was observed by G:Box F3 imaging system (Syngene, Cambridge, UK). The morphologies of GT/siMMP-9 complex were observed on an S-4800 scanning electron microscope (SEM, HI-9056-0003, Hitachi, Japan).
In vitro cytotoxicity of GT/siMMP-9 complex
CCK-8 assay
Cell viability was determined by CCK-8 assay (Dojindo, Japan). HaCaT cells were seeded in 96-well plates (5000 cells/well) in 100 μL of DMEM containing 10% FBS for 24 h. The medium was then replaced with 100 μL of opti-MEM containing GT/siMMP-9, followed by a 24 h incubation. 20 μL of CCK-8 was added in each well and kept for an additional 2 h. Absorbance at 450 nm was measured using the Tecan Spark 10M (Tecan, Shanghai, China) spectrophotometer. Cells without treatment were used as the negative control group, and solutions containing DMEM and CCK-8 without cells was used as blanks. Cell viability (%) = (A450-sample − A450-blank)/ (A450-control − A450-blank) × 100%.
Cell death assay
Cell death was determined by CytoTox96 Nonradioactive Cytotoxicity assay (Promega, WI, USA) based on the calorimetric detection of the released lactate dehydrogenase (LDH). HaCaT cells were seeded in 96-well plates (5000 cells/well) and was incubated with 100 μL of opti-MEM containing GT/siMMP-9 for 24 h. 50 μL of supernatant was mixed with CytoTox96 and was kept for 30 min. Then, 50 μL of stop solution was added to the supernatant. 490 nm absorbance was recorded with a Tecan Spark 10M spectrophotometer. Cells which were treated with lysis buffer were considered as the positive control group, cells without treatment were used as the negative control group, whereas solutions containing DMEM, CytoTox96 and stop solution without cells were used as the blank group. Cytotoxicity (%) = (A490-sample − A490-blank)/(A490-positive control − A490-blank) × 100%.
Apoptosis assay
Early (Annexin V+, 7-AAD−) and late (Annexin V+, 7-AAD+) stage apoptosis (Annexin V Apoptosis Detection Kit, eBioscience, MA, USA) was detected by flow cytometry. HaCaT cells were plated in 6-well plates (40,000 cells/well) with 2 mL of DMEM containing 10% FBS for 24 h. Then, the medium was replaced with opti-MEM containing GT/siMMP-9 and cultured for another 24 h. Cells were harvested, washed twice with PBS, resuspended in 1× Binding Buffer, stained with eFluor 450 conjugated Annexin V and 7-AAD for 15 min at room temperature in the dark, and then analyzed by BD FACSCelesta (BD Biosciences, CA, USA).
In vitro transfection efficiency and lysosomal escape of GT/siRNA
FACS analysis
The cellular uptake of the GT/siFAM complex was evaluated by flow cytometry. HaCaT cells were seeded in 6-well plates (40,000 cells/well) with 2 mL of DMEM containing 10% FBS. After 24 h, the medium was changed into opti-MEM containing GT/siFAM and was further incubated for 4 h. The cells were then rinsed and cultured in fresh medium for 20 h. Cells were collected, washed twice with PBS, resuspended in PBS and subjected to BD FACSCelesta for quantitative analysis. Cells without treatment were considered as the negative control. The efficiency of cellular uptake was represented as the intensity of fluorescein.
Confocal Microscopy analysis (CLSM)
CLSM was used to observe the cellular uptake. HaCaT cells were seeded in 35 mm dishes with glass bottoms (20,000 cells/well) for 24 h. Then, the cells were incubated with opti-MEM containing GT/siCy5 for 4 h, thereafter the medium was replaced with fresh medium and cultured for another 20 h. Cells were washed three times with PBS, fixed with 4% paraformaldehyde (PFA) for 15 min, stained with Hoechst 33342 (1 μg/ml) for 2 min, and imaged with a Zeiss LSM800 confocal microscope (Carl Zeiss Meditec AG, Jena, Germany). Cells without treatment were used as the negative control.
Lysosomal escape
CLSM was also used to observe the lysosomal escape of siCy5. HaCaT cells were seeded in 35 mm dishes with glass bottoms (20,000 cells/well) for 24 h. Then, the cells were stained with LysoTracker Green (KeyGen, Nanjing, China, 1:1000) for 1 h, incubated with GT/siCy5 for 4 h, rinsed and incubated in fresh medium for another 8 h. The cells were then fixed with 4% PFA for 15 min, counterstained with Hoechst 33342 (1 μg/ml) for 2 min and observed with the Zeiss LSM800 confocal microscope.
In vitro MMP-9 silencing of GT/siMMP-9
HaCaT cells were seeded in 6-well plates with 2 mL of DMEM containing 10% FBS for 24 h. The medium was then replaced with opti-MEM containing GT/siMMP-9, followed by a 4 h incubation. The cells were washed with PBS and cultured in DMEM containing 10% FBS for another 20 h. Cells without treatment were considered as negative control.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The expression of MMP-9 mRNA was assayed with qRT-PCR. Total RNA was extracted by TRIzol (TAKARA, Japan) according to the manufacturer’s protocols. Reverse transcription of mRNA into cDNA was carried out using the PrimeScript RT Master Mix (TAKARA) according to the manufacturer's protocols. qRT-PCR was performed on a LightCycler 480II (Roche, Switzerland) with TB Green Premix Ex Taq II (TAKARA) and the primers are shown in Table S2. Data were analyzed using LightCycler480 software.
Western Blot analysis
The expression of MMP-9 protein was detected by Western Blot. Cells were lysed in lysis buffer supplemented with a protease inhibitor cocktail. Protein lysates mixed with loading buffer were heated at 95 °C for 5 min and were then loaded onto 10% SDS- PAGE gels. The proteins were separated out by electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked with 5% BSA for 1 h at room temperature, incubated with MMP-9 antibody (1:1000, Abcam, Massachusetts, USA) and β-actin antibody (1:5000, Abcam) overnight at 4 °C, then incubated with HRP-conjugated secondary antibody for 1 h at room temperature. Blots were detected by the enhanced chemiluminescence (ECL) system (Millipore, Germany) and imaged with the Mini Chemi610 Imaging System (Sagecreation, Beijing, China).
Fabrication, optimization and characterization of PM hydrogels
PM hydrogel was made by blending PF with MC. To prepare PM hydrogel at different MC:PF wt. ratio, MC was first dissolved in PBS solution (4 wt. %) at 4 °C, then appropriate amounts of PF (12, 16 or 20 wt. %) was added and kept for 24 h at 4 °C. PM hydrogels were abbreviated as PM1 (12 wt. % PF and 4 wt. % MC), PM2 (16 wt. % PF and 4 wt. % MC), and PM3 (20 wt. % PF and 4 wt. % MC).
The sol-gel transition of the thermosensitive hydrogel was observed at 4 °C and 37 °C. Rheological properties of gels were measured using an ARES/RFS rheometer (HAKE Company, TA, USA), the elastic modulus (G′) and viscous modulus (G″) were monitored at 5% of strain and a frequency of 1 Hz when the temperature was increased from 5 to 40 °C at a heating rate of 2 °C /min. The temperature at the cross point of G′ and G″ was defined as the gelation temperature. Hydrogel morphology was characterized by the S-4800 scanning electron microscope.
Preparation of PM(GT/siRNA) hydrogel
To prepare PM(GT/siRNA) hydrogels, MC and PF were dissolved completely in PBS solution, then mixed with PBS solution containing GT/siRNA complex to form a gel solution. The final concentration of siRNA was 8 μg/mL.
Release profiles of GT/siFAM complex from PM hydrogels
The solution of PM(GT/siFAM) hydrogels were loaded onto the upper compartment of the transwell insert (pore size: 8 μm, Corning, NM, USA) in a 24-well plate. Samples were kept at 37 °C for gelation before 1.5 mL of PBS was added into the lower compartment. 100 μL of PBS with released GT/siFAM from hydrogels was taken out at predetermined intervals. The wells were replenished with an equal volume of PBS. The amount of the released siFAM was measured by a Tecan Spark 10M spectrophotometer (Tecan, Shanghai, China) and calculated according to the standard curve of GT/siFAM. The cumulative release was calculated with the following formula: Cumulative release (%) = (Wreleased-siFAM / Wtotal-siFAM) × 100%, where Wreleased-siFAM represents the weight of released siFAM, and Wtotal-siFAM represents the weight of total siFAM.
In vitro cytotoxicity of hydrogel-released GT/siMMP-9
PM(GT/siMMP-9) hydrogels were prepared in transwell inserts and immersed in 1.5 mL of opti-MEM as described above, opti-MEM with released GT/siMMP-9 was collected at Day-1, Day-7 and Day-14. HaCaT cells were incubated with the released GT/siMMP-9 for 24 h. CCK-8 was then used to assess the cell viability according to the aforementioned protocol.
In vitro uptake and gene silencing of hydrogel-released GT/siRNA
PM(GT/siRNA) hydrogel was prepared in transwell inserts and immersed in 1.5 mL of opti-MEM, the released GT/siRNA was collected at Day-1, Day-4 and Day-7. HaCaT cells were incubated with the released GT/siRNA for 4 h and then with fresh medium for 20 h. The cellular uptake of the released GT/siFAM was evaluated by flow cytometry. The uptake of the released GT/siCy5 was observed by CLSM. The MMP-9 silencing efficiency of the released GT/siMMP-9 was assessed by qRT-PCR and western blot.
In vivo wound healing experiments
Rats were used for these experiments. Diabetes was induced by a single intraperitoneal (i.p.) injection of freshly dissolved Streptozocin (Sigma-Aldrich, Darmstadt, Germany) at a dosage of 55 mg/kg. After 72 h, blood from the tail vein was collected to examine the blood glucose level with a blood glucose meter (Roche, Switzerland). Rats with a blood glucose reading of over 16.7 mmol/L were considered diabetic.
Four weeks after the injection of Streptozocin, rats were anesthetized with isoflurane (confirmed by toe pinch) and the hair on their backs were shaved. After disinfection with alcohol swabs, full-thickness wounds were created on the dorsal surface using a 10 mm punch biopsy without hurting the underlying muscle. Rats were divided into five groups (n = 6) as the following: (i) non-diabetic rats treated with PBS, (ii) Diabetic rats treated with PBS, (iii) diabetic rats treated with GT/siMMP-9 solution, (iv) diabetic rats treated with PM hydrogel or (v) diabetic rats treated with PM(GT/siMMP-9) hydrogel. Wounds were covered with sterile gauze and bandage, and the rats were individually caged. A ruler was placed beside each wound to record the wound size and were photographed with a digital camera at Day-0, Day-4 and Day-7 after the procedure. All rats were anesthetized at Day-7 post-wounding, the whole wound tissues with a margin of approximately 4 mm of ambient skin were excised, and then sectioned vertically for H&E staining. The wound closure was then calculated with Image J software with the following formula: Wound closure (%) = [(wound areaDay-0 − wound areaDay-7) / wound areaDay 0] × 100%.
In vivo evaluation of MMP-9 expression
Excised skin tissues of non-diabetic rats and diabetic rats that were obtained at Day-0 and Day-7 were evaluated for MMP-9 expression by qRT-PCR, Western Blots and Immunohistochemistry (IHC). IHC was performed on formalin-fixed paraffin-embedded skin sections. The slices were heated at 60 °C for 1 h, washed with xylene and alcohol, blocked with goat serum for 10 min, incubated with MMP-9 antibody for 16 h and peroxidase-labeled polymer for 30 min, then stained with DAB + substrate-chromogen solution and hematoxylin. The whole slides were scanned with a Pannoramic MIDI (3D Histech, Budapest, Hungary), then scored by Quant center software based on the proportion of the positive pixels (dark brown as strong positive, light brown as moderate positive, yellow as weak positive). Histochemistry score (H-SCORE) was calculated with the following formula: H-SCORE = Pweak intensity×1+Pmoderate intensity×2+Pstrong intensity×3, where P represents the percentage of cells.
Evaluation of collagen in skin wound
To examine the collagen of wound tissues at Day-7 after wounding, Masson’s trichrome stain were performed on the wound sections. To further evaluate the collagen type Ⅰ and collagen type Ⅲ, sections were stained with picrosirius red and viewed under a polarized light microscope (Nikon ECLIPSE, Nikon, Japan). The collagen contents were determined by ImageJ software.
In vivo toxicity
At Day-7 after wounding, the rats were anesthetized and their major organs including the heart, lung, liver, kidneys and spleen were collected. These were then fixed with 4% PFA and embedded in paraffin. Tissue sections were stained with H&E and viewed under a Nikon Ni-U optical microscope (Nikon). Blood from the inferior vena cava was collected, and aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN) and creatinine (Cr) values of the serum were measured with an automatic biochemical analyzer (Siemens, Germany).
In vivo biodistribution (BioD)
Diabetic rats were randomly divided into three groups (n = 3), then wounds were created using the protocols mentioned above. These rats were then treated with either (i) PBS, (ii) GT/siCy5 solution, or (iii) PM(GT/siCy5) hydrogel. At Day-1, Day-4 and Day-7 after wounding, the rats were imaged using an in vivo imaging system (NightOWL II LB983, Berthold, Germany), the distribution and intensity of fluorescence around the wounds were recorded and measured. At the same time, the fluorescent images of the major organs including the heart, lung, liver, kidneys, spleen and muscle were also recorded. To further observe the uptake of NPs, wound tissues with a margin of approximately 3 mm of surrounding skin were excised at Day-1, Day-4 and Day-7, frozen sectioned on a sliding microtome to a thickness of 8 μm vertically, and then incubated with Hoechst 33342 for 10 min. The skin sections were observed under a Zeiss Axio Observer.A1 fluorescence microscope.
Statistical analysis
All experiments were performed in triplicate. Data were represented as mean ± standard deviation (SD). Statistical significance was determined by a two-tailed Student’s t-test between two groups, and One-way ANOVA between multiple groups. P value of 0.05 or less was considered statistically significant.
{kind=link}