2.1 Cell culture
The human PANC1 and PaTu8988 pancreatic cancer cell lines, the immortalized pancreatic ductal epithelial cell line H6C7, and human embryonic kidney cell line HEK293T were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were tested and authenticated by short tandem repeat analysis. Cells were cultured in DMEM medium (HyClone, Beijing, China) that contains 10% fetal bovine serum, 1% penicillin and streptomycin, Hyclone, South Logan, UT, USA) and in a humidified incubator at 37°C and 5% CO2 under hypoxia (1% O2) or normal (21% O2) conditions. The plates with adherent cells were put in closed containers full of mixed gas (1% O2, 5% CO2 and 94% N2) for hypoxia condition.
2.2 Antibodies and chemicals
In this study, the antibodies used were rabbit anti-HIF-1α (Cell Signaling, CAT 3716), rabbit anti-OCT4 (Cell Signaling, CAT 2750), rabbit anti-NANOG (Cell Signaling, CAT 4903), rabbit anti-SOX2 (Cell Signaling, CAT 3579), mouse anti-CD44 (Cell Signaling, CAT 5640), rabbit anti-CD133 (Cell Signaling, CAT 64326), rabbit anti-β-Tubulin (Abcam, CAT 21058), rabbit anti-CD9 (Cell Signaling, CAT 13174), rabbit anti-CD63 (Abcam, CAT 68418), rabbit anti-CD81 (Cell Signaling, CAT 56039), rabbit anti-Bax (Cell Signaling, CAT 2772), rabbit anti-Bcl-2 (Cell Signaling, CAT 4223), mouse anti-Caspase-3 (Cell Signaling, CAT 9668), mouse anti-Caspase-9 (Cell Signaling, CAT 9504), Hippo Signaling Antibody Sampler Kit (Cell Signaling, CAT 8579). The chemical was gemcitabine (MedChem Express, HY-17026).
2.3 Western blotting
Cells were rinsed with cold PBS, and then lysed with RIPA buffer containing 1% PMSF, 1% protease inhibitor, 5% 2-mercaptoethanol and 93% 2×loading buffer. The cell lysate boiled at 100°C for 10 min was centrifuged at 12 000g for 10 min. Protein concentration was tested by the BCA assay. Subsequently, the total protein were separated by SDS-PAGE and transferred onto PVDF membranes for immunoblot assays. Membranes were blocked with 5% BSA for 1 h at room temperature and incubated with primary antibodies overnight at 4°C. The membranes washed incubated with the respective HRP-conjugated secondary antibody for 1 h at room temperature, and then visualized with ECL detection system (Amersham Pharmacia Biotech, Little Chalfont, UK).
2.4 CCK-8 assay
4×103 cells per 100µL were seeded into 96-well plates, cultured for 24 hours, and then treated with gemcitabine (0.15, 0.3, 0.6, 1.2, 2.4, 1, 2, 4, 8, 16µM Gem) for 72 hours. After the supernatant was removed, cells were incubated with 10 µL CCK8 reagent without light for 2 hours at 37°C. The absorbance at 450 nm was measured on a microplate reader (Bio-Rad, Hercules, CA, USA). Each experiment was performed in triplicate and the results were analyzed via GraphPad Prism version 7.
2.5 Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted via RNAiso Plus (Invitrogen) according to the manufacturer’s protocol. RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) was applied for synthesizing complementary DNAs (cDNAs) from RNA samples (1 µg). A SYBR Green Mix kit (Bio Rad Laboratories, Hercules, CA) was used to performed qRT-PCR. The relative expression was analyzed based on 2−ΔΔCt method. The primers were designed as Table 1.
Table 1
DNA and RNA nucleotide sequences
|
U6-F
|
CTCGCTTCGGCAGCACA
|
|
U6-R
|
AACGCTTCACGAATTTGCGT
|
|
ROR-F
|
TGCTCCGTGAGAAAGATCCA
|
|
ROR-R
|
GCCGCTAAGCCAAGAAGATC
|
|
OCT4-F
|
CTTGAATCCCGAATGGAAAGGG
|
|
OCT4-R
|
CCTTCCCAAATAGAACCCCCA
|
|
NANOG-F
|
CCCCAGCCTTTACTCTTCCTA
|
|
NANOG-R
|
CCAGGTTGAATTGTTCCAGGTC
|
|
SOX2-F
|
TACAGCATGTCCTACTCGCAG
|
|
SOX2-R
|
GAGGAAGAGGTAACCACAGGG
|
|
CD44-F
|
CTGCCGCTTTGCAGGTGTA
|
|
CD44-R
|
CATTGTGGGCAAGGTGCTATT
|
|
CD133-F
|
GGCCCAGTACAACACTACCAA
|
|
CD133-R
|
ATTCCGCCTCCTAGCACTGAA
|
|
GAPDH-F
|
TGGGGAAGGTGAAGGTCGG
|
|
GAPDH-R
|
CTGGAAGATGGTGATGGGA
|
|
shEGFP-F
|
CCGGTACAACAGCCACAACGTCTATCTCGAGATAGACGTTGTGGCTGTTGTATTTTTG
|
|
shEGFP-R
|
AATTCAAAAATACAACAGCCACAACGTCTATCTCGAGATAGACGTTGTGGCTGTTGTA
|
2.6 Exosome isolation and purification
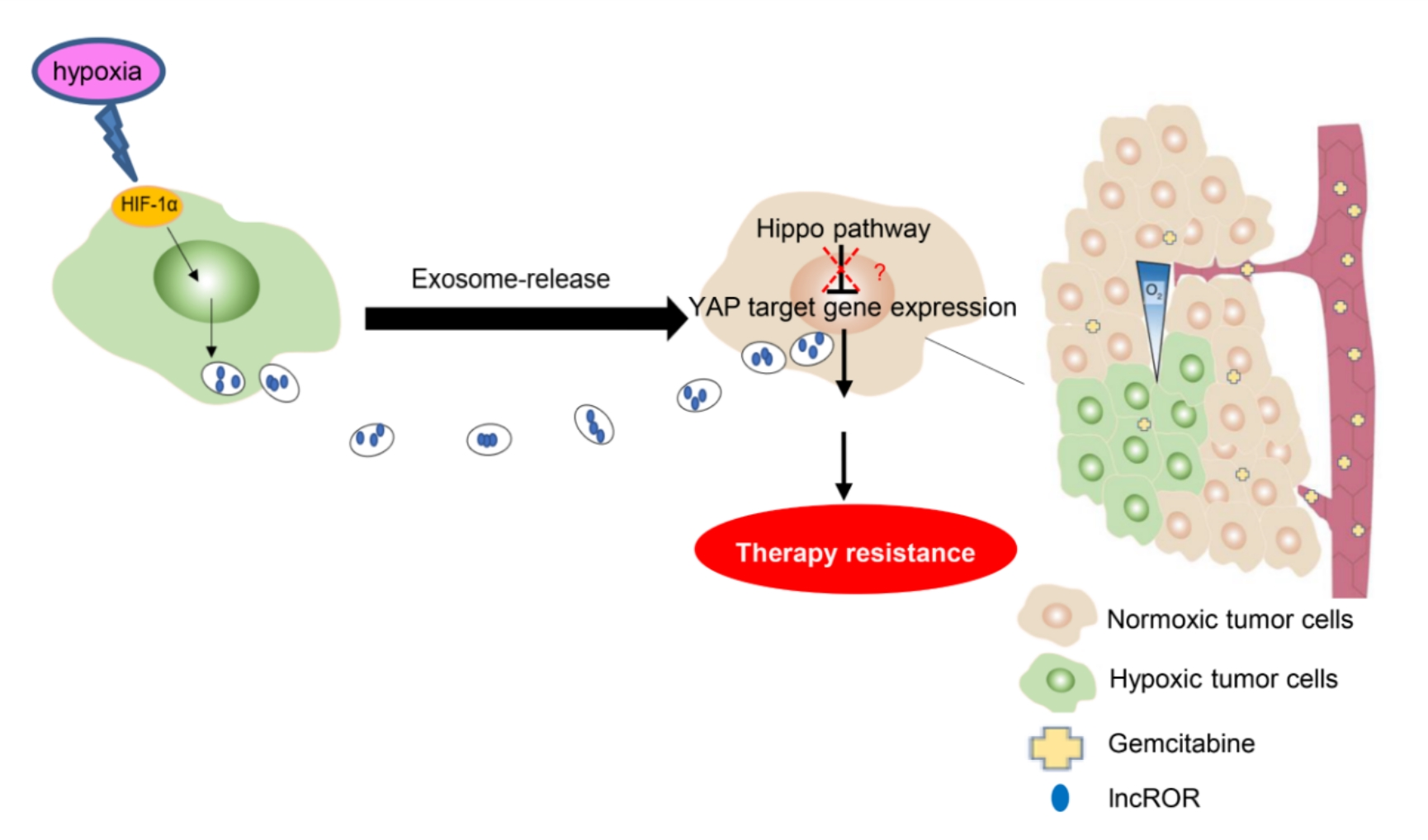
Exosomes were isolated by an exosome extraction kit (EXOTC50A-1, systembio, USA). In brief, cells were cultured in DMEM containing 10% exosome‑depleted FBS (undergoing 100 000 × g ultracentrifugation for 16h). The conditioned medium was collected by 300×g centrifugation for 10 min after 48h treatment. Subsequently, the supernatant was centrifuged at 2 000 × g for 30 min and at 12 000 × g for 30 min. Then exosomes were extracted from the supernatant according to the manufacturer’s protocols. Exosomes resuspended in PBS were purified through a 0.22 µm filter and analyzed by transmission electron microscopy (TEM). Exosome size distribution was calculated using Nanoparticle tracking analysis (NTA). In addition, exosome-specific proteins (CD9, CD63 and CD81) were identified via western blotting. The exosome concentration was evaluated using a bicinchoninic acid (BCA) protein kit (Beyotime, Shanghai, China). For exosome treatment, the cells were cultured with purified exosomes at 100 µg/mL unless otherwise specified.
2.7 Exosome labeling and tracking
Exosomes were labeled using the Dio Green Fluorescent membrane linker dye (Thermo, USA) according to the manufacturer’s instructions. Next, for exosome uptake assay, the traced exosomes were incubated with PANC1 cells for 48 h at 37°C with 5% CO2. After incubation, PBS was used to wash cells and 4′, 6-diamidino-2-phenylindole (DAPI) (Leagene, Beijing, China) was used to stain nuclei for 10 min at room temperature. Finally, Images were captured using an Axio-Imager-LSM800 laser scanning microscope (ZEISS, Oberkohen, Germany). The experiment was performed without light.
2.8 Colony formation assay
Cells were treated with PBS, normoxia exosomes (Nexo) or hypoxia exosomes (Hexo) for 48 hours. After incubation, 1×103 cells each hole were seeded in 6-well plates and maintained at 37°C and 5% CO2 for 10 ~ 14 days. Finally, colonies were fixed in 4% paraformaldehyde and stained with 0.5% crystal violet (Leagene, Beijing, China) for 30 min. The number of spheres formed was counted by Image J.
2.9 Sphere formation assay
2×103 PANC1 cells co-cultured with exosomes for 48h each well were seeded into 6-well plates in serum-free DMEM-F12 supplemented with 20 ng/mL EGF (Beyotime, Shanghai, China), 20 ng/mL bFGF (Beyotime, Shanghai, China) and 1×B27 (Gibco, USA). And the plate was maintained at 37°C and 5% CO2 until spheroids were formed. Then the number of spheres > 50 µm was calculated and plotted.
2.10 Apoptosis Assay
Firstly, PANC1 cells were treated with PBS, normoxia exosomes (Nexo) or hypoxia exosomes (Hexo) for 48 hours. Secondly, 1×105 cells were co-cultured with PBS, Nexo (200µg/well) or Hexo (200µg/well) in DMEM containing 10% Exo-free FBS in 6-well plates for 24 h. Thirdly, gemcitabine was added to the plate at the concentration of 16µM for 72h without light in a humidified incubator. Then 1×105 cells were counted and stained with the Annexin V-APC/PI kit (Fcmacs, Nanjing, China) according to the manufacturer’s instructions. Finally, the apoptotic cells were examined through BD Fortessa FACS (Lake Franklin, NJ, USA) and the apoptosis rate was calculated by FlowJo software 7.6.
2.11 Cell Cycle Analysis
8×105 cells treated with PBS, 100µg/mL Nexo or 100µg/mL Hexo for 48 hours were co-cultured with PBS, Nexo (200µg/well) or Hexo(200µg/well) in DMEM containing 10% Exo-free FBS in 6-well plates for 24 h. Subsequently, PBS or 16µM gemcitabine was added to the plate for 72h without light. Cells were harvested and fixed in 70% cold ethanol for 24 h. Then, according to the manufacturer’s instructions, cells were stained with the Cell Cycle staining Kit (MultiSciences, Hangzhou, China). Samples were detected with a FACS machine, and ModfitLT 5 was used to analyze cell cycle distribution.
2.12 Cell transfection and viral infection
The plasmids pCDH RoR and pCDH Vector were provided by Basic Medical Science Institute, Jiangsu University. DNA sequencing was used to confirm the sequence. Transient transfection was performed using Lipfectamine 2000 regent (Invitrogen, Carlsbad, CA, USA) following the protocols. Plasmid shNC or shROR was co-transfected with psPAX2 and pMD2.G into 293T cells for 48h and 72h, viral particles were collected at centrifugation from the supernatant. PANC1 cells were infected with the virus in DMEM containing polybrene (8 mg/ml, Sigma-Aldrich). After 48h, the culture medium including Puromycin was replaced to select infected cells.
2.13 Statistical analysis
Each experiment was performed separately at least three times. The data was presented as the mean ± standard deviation (SD). Student’s t-test determined the statistical significance between the two groups, while a two-way analysis of variance (ANOVA) was used to analyze the data more than 3 groups. GraphPad Prism 8 software was used to present the results. The statistical significance of the comparison between the two or three groups was analyzed using Student’s t-test or one-way analysis of variance. Statistical significance was considered at P < 0.05.
{kind=link}