Human subjects research was conducted as approved by the institutional review boards of recruiting sites and in accordance with ethical principles laid forth in the Declaration of Helsinki, as previously detailed [10, 20,11]. Only de-identified clinical data and publicly available gene-expression datasets were used for the purposes of this study. Phenotyping data of organ dysfunction trajectories based on clinical and laboratory data available between day 1 through 7 of pediatric intensive care unit (PICU) admission was available in the derivation cohort, as previously detailed [22]. The primary comparison of interest was persistent MODS (death by day 7, persistence of ≥ 2 organ dysfunctions on day 7, or new MODS between days 1-7), relative to those with MODS resolution (≥ 2 organ dysfunctions on day 1 or 3 with < 2 dysfunctions by day 3 and 7 respectively), and septic, non-septic patients with SIRS, and healthy controls. Our choice for this outcome was guided by the fact that patients with death or persistent organ dysfunctions, despite intensive organ support, are likely to represent a subset of patients who may benefit from targeted therapeutic approaches based on their underlying biological predisposition. We conducted analyses with and without inclusion of patients who died within the first 7 days, to test the premise that non-survivors may have a different expression signature relative to survivors with persistent organ dysfunctions. Secondary comparisons focused on those with and without cardiovascular, respiratory, and kidney dysfunction on day 3 and 7 respectively.
Statistical analysis: Demographic and clinical data were summarized with counts and percentages or medians with interquartile ranges. Differences between groups were determined by χ2 test for categorical variables and by one-way analysis of variance (ANOVA) for continuous variables. Dunn’s test was used for account for multiple comparisons testing, where applicable. Log rank test was used to compare 28-day survival among patient subclasses. Logistic regression analyses were used to test the association between MODS endotypes, receipt of adjuvant steroids, and 28-day mortality and occurrence of MODS. All models included the interaction variable between endotype X receipt of steroids. A p-value of 0.05 was used to test statistical significance for all analyses.
Propensity matching: Given the substantial demographic heterogeneity, we generated a propensity score for each patient to account for the confounding influence of age and illness severity, as determined by the PRISM III score [23] for the risk of MODS. We randomly imputed values of 0, 3, and 5 for PRISM-III scores for controls, in whom these data were not available. R package “MatchIt” was used to perform matching and full propensity match method was used. Each patient received a propensity score, which was used to train the machine learning (ML) models and incorporated into risk prediction models.
Gene-expression data: Microarray dataset GSE66099 [24] was downloaded from the NCBI Gene Expression Omnibus (GEO) repository [20] and served as the derivation cohort. The Affymetrix probes were matched to gene symbols using the Affymetrix Human Genome U133 Plus 2.0 (hgu133plus2.db). Data were pre-processed including batch correction for year of study and are detailed online supplement, tables 1 and 2 and figure 1. Differential expression of genes (DEGs) based on a log fold change ≥ ± 0.5, adjusted value for Benjamini Hochberg correction for false discovery rate <0.05, was performed using the limma package [25] in R. We used clusterProfiler [26] for functional gene enrichment, REACTOME pathway analyses [27] to visualize enriched biological pathways, and CIBERSORT [28] to estimate abundance of various immune cells subsets.
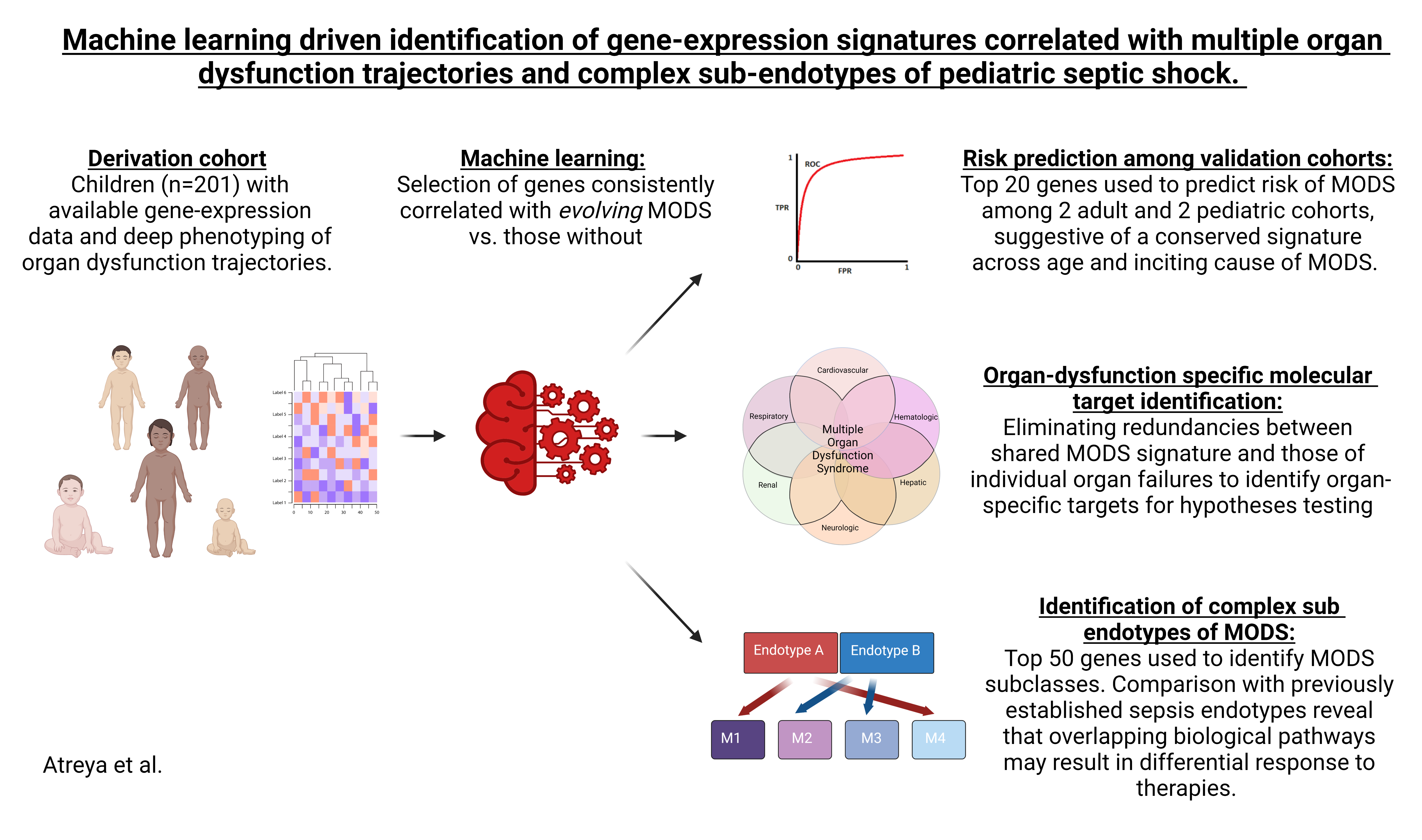
Feature selection: Due to the high dimensionality of the training dataset, we used different feature selection strategies to extract a small number of highly discriminative genes to distinguish patients with MODS, relative to those without. We used three variable selection techniques including Random forests, LASSO, and Minimum redundancy and maximum relevance (MRMR). The genes selected by each of the above methods were aggregated into a single input feature set, and the list of DEGs obtained were added to the list. The propensity score for each patient was included in the list of features used to train the classifier.
Classification models: To counter the imbalance in our training data, we incorporated both undersampling and oversampling techniques into our supervised learning framework, detailed in the online supplement. Briefly, three binary classifications algorithms were used including logistic regression and two tree-based classifiers (Random Forest and Extra Trees classifiers).
We applied a 5-fold cross-validation process that was similar to those previously published by our group [20], and involved randomly partitioning the dataset into five equal subsets in a stratified fashion. Four out of the five subsets formed the training set, and the remaining subset was used for testing set and the process was repeated until each fold had been evaluated as a test data. In each training phase, we first integrated the features obtained using the three feature selection approaches, balanced the dataset using sampling techniques, and finally applied the recursive feature elimination algorithm to arrive at a list of features that were most relevant in predicting the target variable, as summarized in online supplement figure 2. Hyper-parameter tuning was done using a cross-validated grid search technique on a subset of the training data over a parameter grid using the area under the curve as the scoring function. We experimented with different classification thresholds from 0 to 1 with step sizes of 0.001 and chose the one that provided the maximum area under the receiver operator characteristic curve (AUROC). The trained classifier was then used to obtain prediction scores on the hold-out test set. To evaluate robustness of the model training, the entire process was repeated seven times, resulting in thirty-five unique train and test splits. The performances obtained during each run were averaged, and the mean scores along with the 95% CI were reported. Features that were repeatedly chosen (≥ 80% for MODS and ≥60% for individual organ dysfunctions) during multiple runs of the cross-validation experiments were determined. Classification performance of models in external validation cohorts were judged based on the AUROC and the Matthew’s Correlation Coefficient (MCC) – a balanced statistical measure of true positive, true negative, false positive, and false negatives [29], as shown in online supplement figure 3.
External validation: We used 4 external datasets: 1) E-MTAB-5882 ArrayExpress dataset that consisted of time-course-based gene-expression profiling measurements collected from the whole blood of 70 critically injured adult patients in the hyperacute time period within 2 hours of trauma [30], 2) E-MTAB-1548 ArrayExpress dataset comprised of 155 adult post-surgical patients with and without septic shock admitted to a Spanish ICU [31,32]. 3) GSE144406 GEO dataset that consisted of a whole blood bulk RNA sequencing total of 27 pediatric patients including 4 healthy controls, 17 patients with MODS, and 6 patients with MODS requiring extracorporeal membrane oxygen (ECMO) support [33], and 4) E-MTAB-10938 ArrayExpress dataset that consisted of 32 pediatric patients with septic shock, of whom 19 had an immunoparalysis phenotype of MODS [34]. Similar, quality control measures were used during data pre-processing of validation cohorts as with the derivation cohort. Different combinations of top genes (n=10, 20, …50) correlated with MODS in the derivation cohort were tested with numerous classifier and sampling techniques to estimate risk of MODS in validation cohorts. We then determined the minimal number of genes and a single classifier combination that provided consistent performance across validation cohorts. Model performance at a fixed sensitivity of 85% [35] were reported across validation cohorts.
Organ-specific dysfunction signatures: We determined gene signatures that correlated with three major organ dysfunctions — cardiovascular, respiratory, and kidney dysfunction—at day 3 and day 7 time points independently in the derivation cohort. Based on the presumption that the MODS signature represented the shared biological pathways among patients with ≥ 2 of cardiovascular, respiratory, renal, hepatic, hematologic, and neurologic dysfunctions, we identified organ-specific differentially expressed genes by eliminating redundancies associated with the shared MODS signature. Finally, we identified targets correlated with cardiovascular, respiratory, and renal dysfunction which features in ≥ 60% of cross-validation experiments.
Endotype identification: Unsupervised hierarchical clustering of the top genes correlated with persistent MODS, selected based on best stability score [36], were used to derive patient subclasses within the derivation cohort. Clinical relevance of newly derived subclasses was determined by estimating differences in clinical outcomes, organ support, and response to adjuvant corticosteroid therapy. Finally, comparisons between previously validated septic shock endotypes [12] —A and B— available in patients with septic shock and the newly derived MODS subclasses were made. Reactome pathway analyses was used to the determine implicated biological processes [27]. Differences in 50 genes used to determine patient subclasses were compared between MODS endotypes.
{kind=link}