Characteristics of Pholidota plastomes
The graphical genome maps of the newly sequenced Pholidota cp genomes were provided in Fig. 1, generated using OGDRAW [13] and in Additional File 1: Fig. S1, using the GView server [14], respectively. All of the cp genomes exhibited a double-stranded circular quadripartite structure, comprising a large single copy region (LSC; 86,822 bp–87,756 bp), a small single copy region (SSC; 18,598 bp–18,851bp), separated by a pair of inverted repeat regions (IRa and IRb; 26,470 bp–26,721 bp). The thirteen cp genomes ranged in size from 158,786 bp to 159,781 bp (Table 1). For each assembled cp genome, 135 genes were annotated, including 89 protein-coding genes, 38 tRNA genes and eight rRNA genes. The LSC region possessed 60 protein-coding and 21 tRNA genes, whereas, the SSC region only contained ten protein-coding and one tRNA genes. The overall GC content in these plastomes was similar, ranging from 37.27–37.47% and varied within the LSC, SSC and IR regions. The GC content in the IR regions (43.25–43.37%) was higher than those in the LSC (35.14–35.38%) and SSC (30.18–30.47%) regions (Fig. S1; Table 1). Within the IR regions, eight protein-coding genes (rpl2, rpl23, rps7, rps12, rps19, ycf2, ycf15, and ndhB), four rRNA genes (rrn16, rrn23, rrn4.5 and rrn5), and eight tRNA genes (trnA-UGC, trnH-GUG, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC) were present in two copies. In all the plastomes of these species, ycf1 gene was found to extend from IRa into the SSC region, and left a truncated copy at the junction of IRb/SSC. The rps12 gene in Pholidota plastomes was arranged in a trans-spliced state, with 5’-end exon located in the LSC region and two 3’-end exons located in IR regions. Among all identified genes, eleven protein-coding genes (atpF, ndhA, two ndhB, petB, petD, rpl2, rpl16, rpoC1, rps12 and rps16) and eight tRNA genes (two trnA-UGC, trnG-UCC, two trnI-GAU, trnK-UUU, trnL-UAA and trnV-UAC) each contained two exons, while the other four protein-coding genes (two rps12, clpP1 and paf1) each contained three exons.
Table 1
Characteristics of chloroplast genomes of thirteen Pholidota species.
| Species | GenBank accession No. | Genome size (bp) | LSC | SSC | IR | GC (%) | CDS | tRNA | rRNA |
| length (bp) | GC (%) | length (bp) | GC (%) | length (bp) | GC (%) |
| P. articulata | ON880551 | 159,781 | 87,756 | 35.31 | 18,851 | 30.45 | 26,587 | 43.3 | 37.4 | 89 | 38 | 8 |
| P. cantonensis | ON880552 | 158,786 | 86,996 | 35.38 | 18,762 | 30.47 | 26,514 | 43.37 | 37.47 | 89 | 38 | 8 |
| P. chinensis | ON880553 | 159,122 | 86,905 | 35.34 | 18,809 | 30.34 | 26,704 | 43.27 | 37.41 | 89 | 38 | 8 |
| P. imbricata | ON880554 | 159,238 | 87,454 | 35.32 | 18,806 | 30.32 | 26,489 | 43.31 | 37.39 | 89 | 38 | 8 |
| P. leveilleana | ON880555 | 159,353 | 87,321 | 35.29 | 18,734 | 30.43 | 26,649 | 43.28 | 37.39 | 89 | 38 | 8 |
| P. longipes | ON880556 | 158,927 | 86,822 | 35.25 | 18,791 | 30.18 | 26,657 | 43.25 | 37.34 | 89 | 38 | 8 |
| P. missionariorum | ON880557 | 159,029 | 87,257 | 35.33 | 18,600 | 30.35 | 26,586 | 43.27 | 37.4 | 89 | 38 | 8 |
| P. niana | ON880558 | 159,078 | 87,206 | 35.14 | 18,730 | 30.19 | 26,571 | 43.27 | 37.27 | 89 | 38 | 8 |
| P. pallida | ON880559 | 159,171 | 87,399 | 35.27 | 18,832 | 30.34 | 26,470 | 43.33 | 37.37 | 89 | 38 | 8 |
| P. protracta | ON880560 | 159,781 | 87,595 | 35.22 | 18,744 | 30.28 | 26,721 | 43.29 | 37.34 | 89 | 38 | 8 |
| P. ventricosa | ON880561 | 159,418 | 87,408 | 35.17 | 18,598 | 30.42 | 26,706 | 43.29 | 37.34 | 89 | 38 | 8 |
| P. wenshanica | ON880562 | 159,070 | 86,942 | 35.28 | 18,834 | 30.4 | 26,647 | 43.28 | 37.38 | 89 | 38 | 8 |
| P. yunnanensis | ON880563 | 159,729 | 87,618 | 35.21 | 18,849 | 30.3 | 26,631 | 43.28 | 37.32 | 89 | 38 | 8 |
Codon Usage And Amino Acid Frequencies
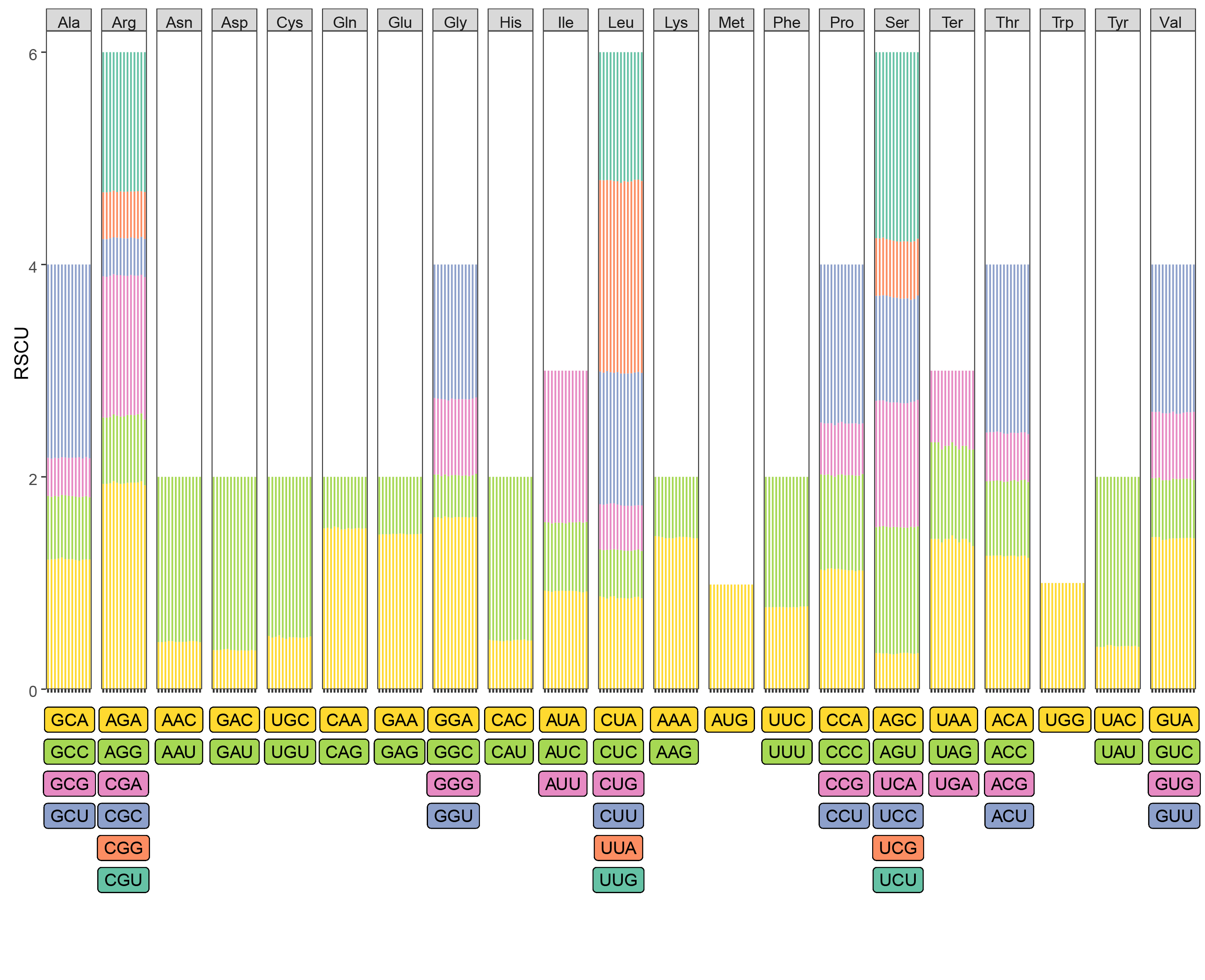
In order to investigate the codon usage pattern, the overall relative synonymous codon usage (RSCU) values of 13 Pholidota cp genomes were calculated (summarized in Additional file 2: Figure S2 and Additional file 3: Table S1). Each cp genome consists of 64 codons with 61 sense codons that code for 21 amino acids (excluding three stop codons, UAA, UAG and UGA). The majority amino acids (19/21, 90.48%) were each encoded by two to six synonymous codons, with the exception of two amino acids methionine (Met) and tryptophan (Trp), which were each encoded by just a single codon (AUG and UGG, respectively). The most frequent amino acids encoded in the plastomes were arginine (Arg), leucine (Leu), and serine (Ser), which were each encoded by six synonymous codons. In contrast, Tryptophan (Trp) was the least common. The RSCU results showed that slightly more than half of the codons (30/59, 50.85%, excluding AUG, UGG and the three stop codons) were more frequently used than expected (RSCU > 1). Almost all of the preferentially used codons (96.67%) ended with A/U except UUG, one of the codons for leucine (Leu). As expected, the use of the start codon AUG and UGG, encoding Trp, exhibited no bias (RSCU = 1). The highest RSCU values (1.92–1.96) were exhibited in AGA encoding Arginine (Arg). The lowest RSCU value at approximately 0.33 was AGC encoding Serine (Ser).
Examination Of Repeats And Ssrs
Repetitive DNA sequences or repeats refer to homologous DNA fragments that occur as a multiple copy of nucleic acids in the genomes, which are a major component of eukaryotic genomes and considered to play an important role in genome stabilization and structural variation [15, 16]. In the current study, we employed REPuter and Tandem Repeats Finder to analyze the repeat sequences in the thirteen cp genomes of Pholidota species. In total, 955 repeats of at least 30 bp long per repeat unit were detected. We categorized these repeats into three types: tandem, dispersed and palindromic. Totally, our repeat analysis identified 444 tandem repeats, 322 palindromic repeats and 189 dispersed repeats in the cp genomes (Fig. 2A; Additional file 4: Table S2). The numbers of tandem repeats varied from 25 to 40; 17 to 33 for palindromic repeats and 7 to 18 for dispersed repeats. Generally, tandem repeats were found as the most prevalent type of repeats, with a proportion of 46.49%, followed by palindromic repeats (33.72%), whereas dispersed repeats were the least common and occupied the lowest portion of 19.79%. In particular, these repeats showed species-specific across the cp genomes. The lengths of dispersed and palindromic repeats varied from 39 bp to 70 bp with the longest repeats presented in P. protracta. The maximum number (40) and minimum number (25) of tandem repeats were detected in P. missionariorum and P. chinensis, respectively. Meanwhile, P. ventricosa cp genome had the highest frequency of palindromic repeats (33), whereas the lowest palindromic repeats (17) were detected in P. cantonensis cp genome. Tandem repeats were found to be highly abundant and frequently occur in these genomes. As shown in Fig. 2A and Additional file 5: Table S3, more than half of the tandem repeats (59.46%) were localized in the LSC region, followed by IR regions (24.77%). The SSC region incorporates the least number of tandem repeats (15.77%) in the genome. Compared to the protein-coding regions (CDS, 23.2%), the intergenic spacer (IGS) regions harbored considerably more tandem repeats (67.57%).
Simple sequence repeats (SSRs), also known as microsatellites or short tandem repeats, refer to short tandemly repetitions of 1–6 base pairs. The SSR analysis of the Pholidota chloroplast genomes using the Perl script MISA detected 525 microsatellites in four main types, namely mono-, di-, tri- and tetra-nucleotide repeats. A comparison of total SSRs for each species is shown in Fig. 2B, with the numbers varied from 32 in P. cantonensis to 48 in P. longipes 32 across the 13 cp genomes (Additional file 6: Table S4). The most abundant SSRs were mono-nucleotides, accounting for approximately 88.38% of all SSRs. Only a small fraction consisted of di-nucleotide (8.76%) and tri-nucleotide (2.67%) repeat motifs. Almost all of the mono-nucleotides contained A /T repeat units (96.77%), with only 3.23% composed of C/G. Meanwhile, all of the di-nucleotides comprised only AT and TA motifs and deficient in CG. Notably, distribution and frequency of different SSR motif types in these cp genomes showed obvious differences. Tri-nucleotide repeats were not present in P. protracta and P. ventricosa. Only one tetra-nucleotide SSR (ATAG) located in the cp genome of P. ventricosa (Fig. 2C; Additional file 6: Table S4). In addition, the identified SSRs were found to be non-uniformly distributed in the chloroplast genomes of the genus Pholidota. The majority of SSRs resided in the LSC region (70.45–82.86%), followed by SSC region (10.64–22.92%), while a minority (4.17–12.5%) occurred within the IR regions (Fig. 2D; Additional file 6: Table S4).
Mining Of Snp And Indel Markers
Single-nucleotide polymorphisms (SNPs) and DNA insertions-deletions (InDels) are useful polymorphic markers for analysis of genetic diversity and genetic mapping. In this survey, we determined these genetic variants based on a comparison of 13 cp genome alignments with the P. longipes cp genome as a reference. In total, 22,464 mutations were identified, including 13,834 SNPs and 8,630 InDels (Additional file 7: Table S5). The number and distribution of SNPs and InDels detected in each species are displayed as bar graphs in different colors (Fig. 3, rings “2–13”). The number and frequency of SNPs and InDels varied considerably across the 13 Pholidota plastomes. The maximum number of SNPs was detected in the P. ventricosa cp genome (1471) and the minimum number was found in the P. niana cp genome (233). Similarly, the maximum number of InDels was also detected in P. ventricosa (916), and the lowest was observed in P. niana (264). The average numbers of SNPs and InDels were 1064 and 267, respectively. Most of Pholidota species have 1,100 to 1,500 SNPs and 280 to 400 InDels. However, the SNPs in the cp genomes of P. niana (233) and P. protracta (391) were significantly fewer than those in the other species. Similarly, fewer than 160 InDels were discovered in P. niana (62) and P. protracta (153) (Table S5). The low number of interspecific polymorphisms revealed high chloroplast sequence similarity among P. longipes, P. niana and P. longipes. Given the lower number of SNPs and InDels detected in P. niana and P. protracta, these species are most closely related to each other.
The frequency and density of SNP/InDel loci identified in the genomic regions varied across the thirteen cp genomes (Additional file 7: Table S5). A large number of variants resided in LSC regions, followed by the SSC region. The lowest number of polymorphic sites was noticed in the IR regions (Fig. 3). In addition, the overall distribution of SNPs and InDels among the cp genomes shared a similar pattern. Sequences in the non-coding regions exhibited significantly higher divergence than that in the coding regions. For the non-coding regions, sequences of the intergenic spacer regions comprise a large majority of variants.
Comparison Of Ir-sc Border Positions
The borders of LSC, SSC and IR regions of thirteen Pholidota cp genomes were compared to determine unique and common features (Fig. 4). In general, these cp genomes exhibited relatively stable patterns with similar gene content and arrangement. The LSC/IRb boundary lied between rpl22 and rps19 genes, while the IRa/LSC border was located between rps19 and psbA genes. Two intact copies of the rps19 gene were present near the IR-LSC borders. In particular, the ycf1 gene crossed the SSC/IRa boundary, leading to an incomplete duplication of this gene at the IRb/SSC border. In all cp genomes sequenced, the partially duplicated ycf1 gene spanned the IRb/SSC border and interlaced with the ndhF gene, extending to various lengths into the SSC region.
Although the length of the IR regions varied little across the thirteen Pholidota cp genomes, ranging from 26,470 bp to 26,721 bp, the IR/SC boundary regions of these species present certain discrepancies. The SSC/IRa junction was situated in the ycf1 coding region, with a size variation from 5,378 bp (P. missionariorum) to 5,588 bp (P. yunnanensis). At the SSC/IRa border, the ycf1 gene extended into the SSC region, at varying lengths ranging from 4,367 bp in P. missionariorum to 4,568 bp in P. yunnanensis. The truncated copy of ycf1 was largely located in the IRb region, with its one end extending into the SSC region, ranging from 7 bp (P. longipes) to 25 bp (P. protracta). In contrast, ndhF was mainly situated in the SSC region, partially overlapping with the duplicated ycf1 gene. The ndhF gene showed the same length of 2,258 bp in all species, whereas the portion located in the IRb region varied in length from 53 bp in P. protracta to 71 bp in P. longipes. The distance between rps19 and LSC/IRb border was 47 bp in P. cantonensis, whereas 167 bp in P. chinensis. The length from rpl22 to the LSC/IRb border was 43 bp in P. articulata and P. chinensis, in contrast to 73 bp in P. leveilleana and P. yunnanensis. On the other side of the IRa/LSC boundary, psbA gene was found in the LSC region of all genomes but was located 84 bp (P. missionariorum) to 109 bp (P. imbricata and P. pallida) apart from the IRa/LSC border.
In general, the Pholidota cp genomes showed obvious changes at the IR/SC boundaries and adjacent genes. Two genes ycf1 and ndhF located at the IR/SSC junctions were found to be highly variable. The results also showed that some differences exist in the non-coding intergenic regions (rpl22-rps19-psbA).
Genome sequence divergence among Pholidota species
The sequence divergence across the thirteen Pholidota cp genomes were compared and plotted using mVISTA by aligning these cp genomes with the annotated P. longipes cp genome as a reference (Fig. 5). The whole-genome alignment revealed that sequence variations in the non-coding regions in orange bars were greater than that in the protein-coding regions (CDS) as colored in purple bars. The two IR regions were more conservative than the LSC and SSC regions. The highly divergent non-coding regions in these cp genomes appeared in the intergenic spacer regions (IGS), such as trnK-UUU-rps16, rps16-trnQ-UUG, trnR-UCU-atpA, atpF-atpH, trnC-GCA-petN, trnS-GGA-rps4, rps4-trnF-GAA, trnT-UGU/trnL-UAA, ndhC-trnV-UAC, atpB-rbcL, accD-psal, clpP1 intron, psbB-psbT, petD-rpoA, rpl16-rps3, rps12-trnV-GAC, ndhF-rpl32, rpl32-trnL-UAG, psaC-ndhE, and trnV-GAC. In CDS regions, some genes, such as psbA, rps16, atpF, atpH, rbcL, psbT, petD, rpl16, rpl32, ycf1 and ycf2 genes showed relatively high variations among these genomes. By contrast, all the rRNA genes were highly conserved when compared to the other genes.
To test divergence level within different regions of these cp genomes, the nucleotide diversity (Pi) was measured by DnaSP within 600-bp windows (Fig. 6). The Pi values for each genome ranged from 0 to 0.1174. Of the protein-coding regions (CDS), the average Pi value was 0.0057. Compared to the coding regions, the intergenic spacer (IGS) regions showed comparably higher divergence levels, with an average Pi value of 0.0092. As expected, variations in the IR regions of the cp genomes were considerably lower than that of SC regions. The SSC region showed the highest nucleotide diversity in view of its average Pi value of 0.0159, followed by LSC region, with an average of 0.0090, whereas the average Pi in the IR region was 0.0020.
We selected six highly variable regions in the cp genomes with a nucleotide variability (Pi) higher than 0.06 (Fig. 6). These regions were four intergenic spacer regions: rps16-trnQ-UUG (0.0790), ndhF-rpl32 (0.0758), rpoB-trnC-GCA (0.0630), rps11-rpl36 (0.0624), and two plastid genes ycf1 (0.1174), ndhA (0.0624) within the coding regions. Among these regions, ndhF-rpl32 was located at the IRb/SSC junction, ycf1 crossed the SSC/IRa border. ndhA was situated in the SSC region, whereas three of six (rps16-trnQ-UUG, rpoB-trnC-GCA and rps11-rpl36) were located in the LSC region (Fig. 6). These divergence hotspots could serve as significant genetic markers for species delimitation and phylogeographic analyses.
Phylogenomic Analysis
To determine the phylogenetic positions of Pholidota species and better clarify their evolutionary relationships, we constructed phylogenetic trees based on the complete plastome sequences of 22 species with maximum likelihood (ML) and Bayesian inference (BI) methods. In addition to the 13 newly sequenced genomes, three published cp genomes of Pholidota and four published cp genomes from other genera Bulleyia, Coelogyne and Panisea were also retrieved for analyses. Two species of genus Pleione were selected as outgroups based on previously reported relationships [17]. The ML and BI phylogenetic analyses yielded consistent topologies. The phylogram of the ML tree with the support values at the nodes is shown in Fig. 7.
The sampled species of Pholidota were generally separated into two main clades, namely clades A and B (Fig. 7). As the earliest differentiated lineage, clade B including an accession of species P. ventricosa, was placed as an unsupported sister to the remaining members of Pholidota. Clade A (PP = 1.0, BP = 100) containing most other sampled Pholidota species was further divided into three well-supported lineages, designated as subclades I, III, and IV that were found each related to the other genera of the subtribe Coelogyninae. Of the three subclades, subclade I consists of P. imbricata and P. pallida, forming the sister clades to P. articulata, and P. chinensis. Subclade II, which includes two sampled species of Coelogyne (C. fimbriata, C. ovalis), was strongly supported as the sister to subclade I (PP = 1.0, BP = 100). Within clade III, an accession of Panisea uniflora, fell within the lineage with accessions of P. longipes, P. niana, and P. protracta, constituting a strongly supported monophyletic group (PP = 1.0, BP = 100), with the former two as the closest sisters. Clade IV consisting of monotypic genus Bulleyia, with its sole representative, B. yunnanensis, together with six accessions of Pholidota, formed a robust monophyletic lineage (PP = 1.0, BP = 100). A small clade with accessions of P. cantonensis and P. missionariorum was placed as successive sisters to the clade with accessions of P. leveilleana, P. wenshanica and two accessions of P. yunnanensis (PP = 1.0, BP = 100).
{kind=link}