A quantitative estimation of the oxidative folding machinery in E. coli
An initial aim in this study was to estimate the required rates of de novo disulfide bond formation and disulfide bond isomerisation in E. coli on the one hand, and the abundance of components of the oxidative folding machinery and the enzymatic activity they provide on the other. We then set out to bring these two elements together by using a quantitative modelling approach to describe the oxidative folding system dynamically.
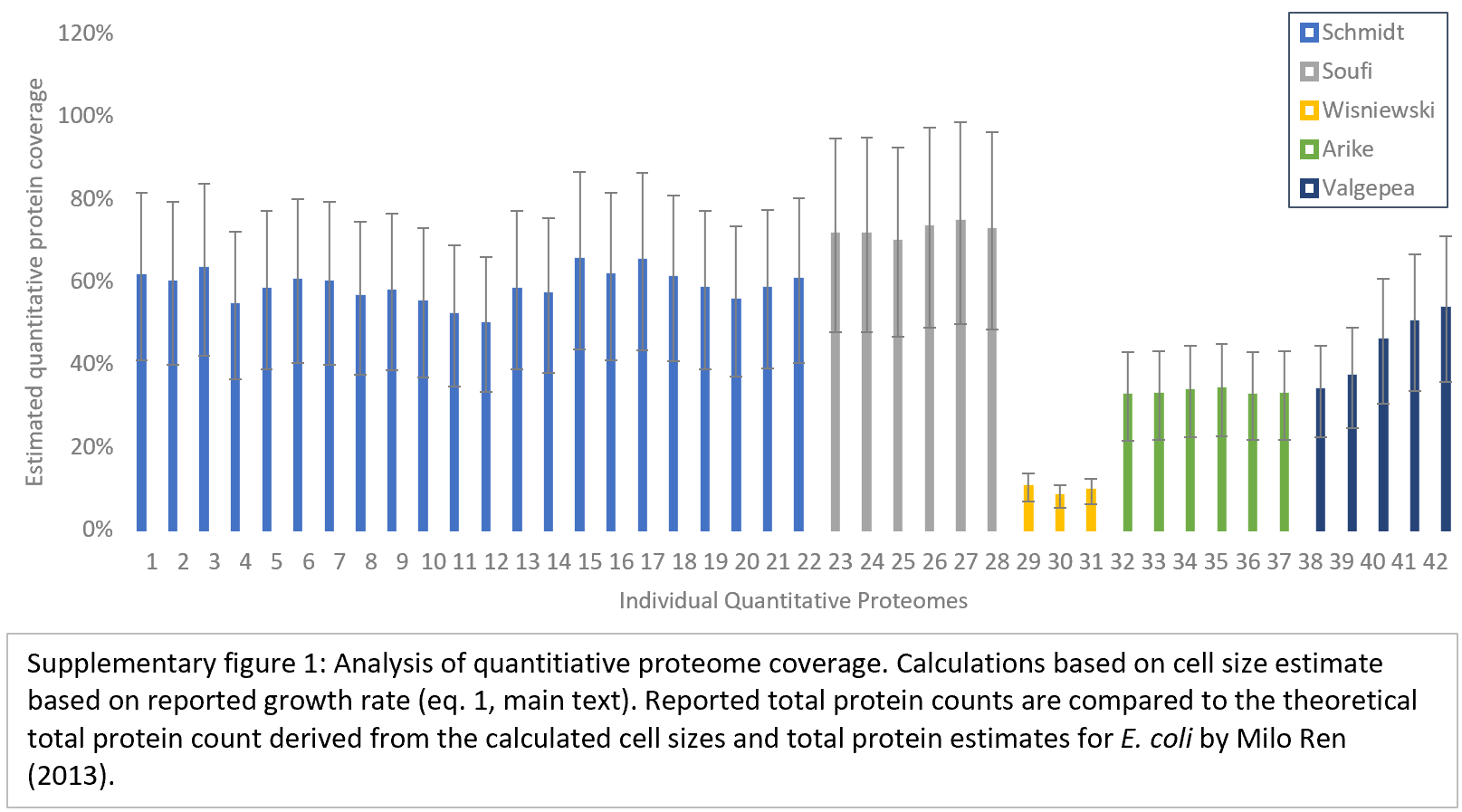
We initially collected a total of 73 quantitative E. coli proteomes from seven different publications (7–13). Six of these publications provide absolute protein quantification values in the form of protein copy numbers per cell. A seventh study by Peebo et al. provides protein concentrations, which we converted to protein copy numbers per cell by estimating the cell size from data provided in the study as explained in the Materials and Methods section. The collected proteomes cover a variety of growth conditions, differing in media composition, carbon sources, growth rates, E.coli strains and stresses imposed on the cells. To select the most suitable datasets for modelling, we analysed the proteomes in terms of their quantitative protein coverage as well as completeness of the supplementary information. In a first step we excluded proteomes without reported growth rates since this information was essential for estimating protein synthesis rates (see below). In order to estimate proteome coverage in these studies, reported values for total cellular protein count were compared to the corresponding theoretical total cellular protein count based on published calculations (14), which exploit the fact that cellular protein count correlates with both cell size and cellular growth rates. Growth rates were used to estimate the corresponding cell sizes using Eq. (1). The resulting estimates of fractional proteome coverage are displayed in supplementary Fig. 1. Proteomes with a quantitative coverage below 50% were not considered for the quantitative modelling part.
Good overall proteome coverage is important for good representation of oxidative folding substrates in the datasets. In addition, we intended to use the datasets also as a source for evaluating the abundance of oxidative folding enzymes, and we therefore specifically investigated how well the dsb enzymes were represented in them.
The primary oxidase dsbA is an abundant, soluble protein detected in all datasets with a mean abundance of 696 ppm (proteins per million proteins) or around 4000 proteins per cell (Fig. 1). The two isomerases dsbC and dsbG are also soluble proteins. The more abundant dsbC was again represented in all datasets with a mean concentration of 144 ppm, or 800–900 proteins per cell. The less abundant dsbG was not represented in two of the datasets, but in those datasets where it was represented the concentration was reported with a mean abundance of 27 ppm or around 160 proteins per cell. In contrast to the good representation of the soluble enzymes, the membrane-bound dsbB and dsbD had no associated abundance data in the majority of studies, only being covered in 25 and 23 of the 73 datasets, respectively. This was expected, since membrane-associated proteins are frequently under-reported in proteomics datasets if not specifically accounted for during sample preparation (15).
Because of the poor representation of the membrane-associated oxidative folding enzymes, we estimated their abundance from synthesis rate data reported by Li et al (16). This study used a ribosome footprinting approach to characterise protein synthesis activity in the E. coli translatome. By determining apparent synthesis rates for all dsb proteins, we were able to establish a relationship between synthesis rates and steady state levels for dsbA, C and G, which we then used to predict steady state levels for dsbB and D from their synthesis rates. These analyses yielded mean concentrations of 139 ppm and 29 ppm for dsbB and dsbD, respectively (corresponding to around 480 and 100 proteins per cell).
Based on the overall quantitative coverage analysis, the availability of supplementary information regarding growth rates, cell size and stress conditions as well as the coverage of the key ‘dsb’ enzymes, the proteomes reported by Schmidt et al. (13) were selected for the rest of this study (unless noted otherwise).
The E. coli disulfide proteome
Following initial quality controls and selection of suitable datasets, we used the proteomics data do estimate the volume of disulfide bonds processed by the dsb enzymes in native E. coli cells. Proteins are substrates if they are located in the periplasm and contain disulfide bonds in their folded state, and we used ancillary data sources to identify the subset of cellular proteins to which these criteria apply.
Three different sources of known disulfides in the E. coli proteome were considered to identify disulfide-bond containing proteins: The Uniprot database (17) which contains curated annotations from the research literature and two proteomics studies (18, 19) which used labelling techniques to identify native E. coli disulfide bonds by mass spectrometry. The three data sets collectively identified 360 distinct disulfide bonds that can form in E. coli proteins (Fig. 2A); however, only 45 of these were identified in all three (Fig. 2B). These initial observations suggest considerable residual uncertainty when it comes to the types of disulfide bond that can form in E. coli proteins. However, agreement between the studies is better for highly expressed proteins, and in consequence the uncertainty regarding the total number of disulfide bonds that are formed in bacterial cells is much lower (Fig. 2B).
To identify proteins located in the periplasm, we used data generated by Loos et al. (20) who trained a machine learning algorithm to annotate protein locations based on a protein’s primary amino acid sequence, which suggests locations for 98% of all known E. coli proteins with high confidence. We considered all proteins annotated in this dataset as ‘secreted’ and ‘secreted outer membrane’ as potential substrates for the dsb machinery. An overview of numbers of periplasmic disulfide bonds identified in this way is presented in Fig. 2.
A closer look at the reported disulfide bonded proteins confirms the common assumption that E. coli has a relatively simple disulfide proteome (Fig. 2A). The total set of 360 reported disulfide bonds are located on 285 different proteins, with only 18 proteins having more than one potential disulfide bond. 174 of the 360 identified disulfide bonds can be allocated to either the periplasm or the outer cell membrane. Of those, 140 are formed by consecutive cysteines, 12 by non-consecutive cysteines and 22 are intramolecular. Out of the 174 disulfides in bona fide periplasmic proteins, 97 are described by 2 + sources, and we used this “higher confidence” subset for the kinetic modelling studies described in the following (Fig. 2B). This set of 97 disulfide bonds is located on 82 individual proteins, with only 6 proteins having more than 2 potential disulfide bonds.
The disulfide bond datasets provide a static picture of disulfide bonds in the E. coli proteome. However, it does not yet incorporate information on the folding pathways by which individual disulfide bonds are formed, and in particular whether correct bonds are formed immediately through the action of dsbA or following initial incorrect formation and subsequent isomerisation by dsbC or dsbG. To our knowledge there is no quantitative, proteome-wide information available to address this question. We therefore introduced a number of semi-quantitative assumptions that we then used to formulate boundary conditions for required levels of isomerase activity in the cell. We categorised disulfide bonds into four categories which we assume are of increasing risk of misfolding, based on relative cystine locations on each protein’s amino acid chain. The first category contains proteins that only have two cysteines, where no mispairing is possible. The second category contains proteins where disulfide bonds are formed between consecutive cysteines, but where additional cysteines exist that could mis-pair with either of the cysteines involved in the native bond. Thirdly we consider disulfide bonds between non-consecutive cysteines, where we assume a more substantial risk of incorrect disulfide bond formation with the intervening cysteine. A fourth category contains intermolecular disulfide bonds and was not further considered in this analysis.
A quantitative evaluation of proteins and associated disulfide bonds in each “folding difficulty” is shown in Fig. 2C. This graph displays total numbers of disulfide bonds in each category, calculated as the number of proteins in each category multiplied with each protein’s abundance. Overall the risk of disulfide-bond related misfolding in E. coli appears relatively low, since two thirds of disulfide bonded proteins contain exactly the two cysteines required for the disulfide bond to form, without any scope for misfolding. This finding is consistent in principle with the observation reported above that the enzymes involved in disulfide bond isomerisation (dsbCDG) are expressed at much lower levels compared to dsbAB.
Modelling oxidative folding and isomerisation
To investigate the dynamics of periplasmic oxidative folding processes in E. coli, we used an ODE-based computational model with a reaction scheme as depicted in Fig. 4. The model assumes a steady influx of folding substrates into the ER, where the substrates are grouped into the different folding categories shown in Fig. 2. The rate of substrate influx into the ER is estimated from the quantitative disulfide proteome and the cells’ growth rate, which gives the minimum rate of protein synthesis required to maintain a stable proteome in the steady state. In reality additional protein synthesis is required to counteract protein turnover, but under rapid growth conditions this proportion is small compared to that required due to growth (21).
In the model, proteins enter the periplasm in a reduced and unfolded state (UF) but can be oxidised by interaction with dsbAO. The outcome of this oxidation can be either the adoption of a misfolded (MFP) or a correctly folded (FP) state. We assume that the reaction kinetics leading from UF to FP and MFP are identical, but that the probabilities of immediately adopting the FP state differ for the three substrate classes, being 100% for ‘cat1’, 50% for ‘cat2’ and 0% for ‘cat3’. Although actual proteins are more likely to form a continuum of folding probabilities, we assume that these discrete protein categories in the model capture the different types of behaviour observed in biological proteomes both in terms of the types of reactions that occur and (in a first approximation) in terms of their quantitative requirements of de novo folding and isomerase activities provided by the E. coli oxidative folding pathways.
Following oxidation of UF proteins, dsbA is reduced and can be regenerated by dsbB, which in turn is regenerated in a redox reaction with quinone. If UF oxidation leads to formation of MFPs, these can further interact with the isomerase dsbCG where one of three things can happen: 1) the disulfide bond is successfully isomerised to form FP, 2) the isomerisation is unsuccessful and the protein remains in the MFP state or 3) the disulfide bond on the substrate is reduced by the isomerase, returning the protein to the UF state. In the latter case, the isomerase itself becomes oxidised and has to be regenerated via dsbD, which in turn transfers the excess electrons onto thioredoxin. In the model representation in Fig. 3, quinones and thioredoxin are depicted in the periplasm for simplicity, even though in reality these reactions take place on the cytoplasmic side of the transmembrane proteins. However, since the model simplifies the reoxidation of dsbB and the reduction of dsbD into pseudo-first order reactions, the actual location of these terminal components is irrelevant in this context.
Each model reaction is represented by an ordinary differential equation and has an associated kinetic parameter. These kinetic parameters dictate the speed of each reaction, and in cases where there is more than one possible reaction outcome, also the ratio between the possible outcomes.
Kinetics
We initially parameterised the model using enzyme concentrations derived from the quantitative proteomics data, and substrate concentrations derived from proteomics data covering disulfide-bonded proteins in the different “folding difficulty” categories outlined above. We assume that clients of the dsb proteins are predominantly newly translated proteins which are not yet correctly folded. The rates with which such dsb clients are generated can be estimated from their cellular abundance in the steady state and from the growth rate, since the rate of growth dilution dominates rates of protein turnover in fast growing microbial cells (22).
Based on these known substrate production rates, we then characterised minimal enzyme rate constants that were compatible with the essential requirement of doubling the E. coli proteome once per generation. This strategy allows estimating minimal required enzyme activities for the core reactions in the model but may underestimate the actually required activity if futile cycles occur frequently. For example, if a disulfide bond formed by dsbA is resolved again by dsbCG, or if a protein in a mis-folded state is simply transferred to another mis-folded state rather than the correctly folded one, enzyme activity is engaged without a net change in substrate or product concentrations. Because we have no information allowing us to estimate the frequency of such cycles, we assumed here that such cycles are rare compared to productive folding events. In our model parameters, we assumed arbitrarily that futile cycles make up one third of all isomerase-catalysed reactions.
We applied this strategy to all datasets generated by Schmidt et al. (13), thus generating specific minimally required enzyme rate constants for each of the growth conditions investigated in this study. Due to the specific reaction structure employed in the model, the results are returned in the form of apparent association rate constants for the formation of enzymes–substrate complexes. It is worth noting the relationship between these reported apparent rate constants and the actual enzyme rate constants: because we characterise minimal rate constants required for the system to cope with the observed substrate influx, these are expected to be slower than actual biochemical enzyme rate constants if an enzyme is not engaged at its maximum capacity. On the other hand, the modelled apparent rate constants cannot be faster than the actual rate constant as this would be biochemically impossible and would indicate that either enzyme or substrate concentrations have been reported incorrectly, or that the model structure has been chosen inappropriately.
Table 1
Reaction rates for oxidative folding in E. coli
| |
|
Reaction(s)
|
| |
R2
|
R3
|
R4 (Cat1)
|
R4,5 (Cat2)
|
R5 (Cat3)
|
R6,7,8 (Cat2)
|
R6,7,8 (Cat3)
|
|
Chemostat
|
Chemostat µ = 0.5
|
0.090
|
5.388
|
0.946
|
0.433
|
1.297
|
0.227
|
0.527
|
|
Chemostat µ = 0.35
|
0.091
|
4.526
|
1.093
|
0.703
|
2.106
|
0.673
|
1.957
|
|
Chemostat µ = 0.20
|
0.040
|
3.943
|
0.791
|
0.535
|
1.603
|
0.295
|
0.886
|
|
Chemostat µ = 0.12
|
0.016
|
3.181
|
1.367
|
0.924
|
2.770
|
0.185
|
0.557
|
|
High Growth
|
LB
|
0.228
|
6.727
|
1.894
|
0.337
|
1.131
|
0.246
|
0.584
|
|
Glycerol + AA
|
0.199
|
6.708
|
2.519
|
0.471
|
1.151
|
0.364
|
0.371
|
|
Stress
|
pH6 glucose
|
0.152
|
7.571
|
1.599
|
0.733
|
2.084
|
0.227
|
0.486
|
|
42°C glucose
|
0.048
|
6.801
|
1.023
|
0.370
|
1.108
|
0.112
|
0.240
|
|
Osmotic-stress glucose
|
0.111
|
6.637
|
1.107
|
0.482
|
1.518
|
0.192
|
0.412
|
|
non-stress, sub-optimal
|
Glycerol
|
0.048
|
5.183
|
0.892
|
0.486
|
1.426
|
0.122
|
0.321
|
|
Glucosamine
|
0.083
|
5.008
|
1.005
|
0.537
|
1.608
|
0.210
|
0.515
|
|
Xylose
|
0.077
|
4.400
|
0.772
|
0.413
|
1.059
|
0.145
|
0.344
|
|
Glucose
|
0.080
|
4.309
|
0.796
|
0.404
|
1.092
|
0.168
|
0.292
|
|
Succinate
|
0.065
|
3.925
|
0.873
|
0.481
|
1.440
|
0.176
|
0.453
|
|
Fumarate
|
0.065
|
3.888
|
0.988
|
0.556
|
1.664
|
0.158
|
0.406
|
|
Mannose
|
0.072
|
3.599
|
0.722
|
0.320
|
0.960
|
0.119
|
0.321
|
|
Galactose
|
0.044
|
3.549
|
0.857
|
0.562
|
1.737
|
0.165
|
0.505
|
|
Pyruvate
|
0.042
|
3.351
|
0.896
|
0.576
|
1.726
|
0.104
|
0.312
|
|
Fructose
|
0.079
|
3.230
|
1.061
|
0.425
|
1.149
|
0.187
|
0.301
|
|
Acetate
|
0.046
|
2.758
|
0.613
|
0.394
|
1.181
|
0.100
|
0.299
|
| |
Median
|
0.07
|
4.35
|
0.97
|
0.48
|
1.43
|
0.18
|
0.41
|
| |
Range (fold)
|
14.3
|
2.7
|
4.1
|
2.9
|
2.9
|
6.7
|
8.2
|
| |
Inter Quartile Range (fold)
|
1.9
|
1.6
|
1.3
|
1.4
|
1.5
|
1.6
|
1.6
|
To facilitate interpretation, we multiplied the enzyme concentrations for each condition with the modelled apparent association rate constants, thereby creating a pseudo first-order rate constant expressing how rapidly substrates are likely to be processed in each of the different growth conditions (Table 1). Lower first-order rate constants indicate that enzymes engage less readily with their substrate, because in the respective condition the ratio of provided to required activity is lower. In terms of the question we initially asked, high-rate constants thus imply a degree of oversupply in the system, which could be exploited for example for more efficient recombinant protein processing.
We observed significant variation in oxidative folding capacity between the different growth conditions (Table 1). The de novo folding reactions (R4,5 in Fig. 3) vary over a two- to four-fold range between conditions, and the isomerisation reactions vary over a six- to eight-fold range. Each of the investigated proteome datasets reflects specific combinations of growth rates, oxidative folding enzymes and their substrates, and the observed folding capacity most likely changes as the result of relative changes in these parameters.
We observed a clear dependence of the capacity for dsbA reoxidation by dsbB with growth conditions. The highest capacities for this reaction, with substrate processing rates of 6 min-1 and higher, were observed during stress conditions which in this dataset included low pH, high temperature and osmotic stress; as well as growth in LB and amino-acid supplemented glycerol, two non-stress conditions with high growth rates. Moreover, the chemostat series of experiments, in which growth rates are directly controlled by the dilution rate with otherwise identical parameters, revealed a strong correlation between the capacity to regenerate dsbA and growth rates (Figure 4A. The Pearson’s Product-Moment Correlation Coefficient for the correlation between growth rate and R3 rate parameters for this reaction is 0.98). None of the other reaction rates show similar patterns, and in particular the de novo folding reactions (R4,5) show no clear correlation with the same conditions. Indeed, the majority of the apparent R4/5 rates appears remarkably constant with the lowest inter-quartile range of all reactions. One interpretation of these findings is that E. coli cells adjust the provision of dsbA, including the cells’ ability to reactivate this enzyme, in line with demand arising from increasing growth speed, thus enabling the timely processing of inflowing substrates.
Interestingly, the maintenance of dsbA capacity under high growth and stress conditions appears to be the result of distinct set-ups of the oxidative folding machinery (Fig. 4). During chemostat growth, the abundance of both dsbA and dsbB increases, resulting in an overall increase in the capacity to regenerate dsbA (Fig. 4A). This scenario is consistent with an increasing need for de novo folding in response to the increased dilution rates during fast growth, when the influx of dsbA substrates increases proportionally with growth and dilution rates. Under such conditions, both dsbA and the capacity to regenerate this enzyme must be adjusted concomitantly. In contrast, the high levels of dsbA under stress conditions appear to be efficiently reoxidised (they have high R3 rates) despite low dsbB concentrations. As the overall capacity of dsbA to catalyse de novo folding is still high under stress conditions compared to normal growth (see apparent R4 rates in Table 1), the most parsimonious explanation is that during stress high dsbA levels are maintained despite relatively low de novo folding demand.
Oxidative folding demand and recombinant protein production capacity
In addition to analysing the native capacity for oxidative folding in E. coli, the model allows estimating the capacity of this organism for dealing with additional substrates such as recombinant proteins. We investigated this by increasing the production rate for oxidative folding substrates of varying “folding difficulty”, i.e., where the probability of immediately adopting the correct fold decreases and the probability of adopting an incorrect fold which needs to be further corrected by an isomerase increase. We assume that there is no regulatory adaption to the new substrate. In order to monitor the capacity to process recombinant substrates under particular growth conditions, we increase the rate with which additional recombinant proteins are produced until the cells’ native substrates begin to accumulate (we use a threshold of 0.5% of substrates accumulating in the unfolded state). The amount of recombinant protein that can be introduced before this threshold is reached is displayed in Fig. 5.
The results suggest that the capacity to produce recombinant protein varies strongly with growth conditions, as well as with the requirement for isomerisation. Growth in glycerol, glucose and pyruvate are predicted to allow the highest yields in principle, with an estimated capacity of processing up to 150% of recombinant protein over and above the normal cellular protein complement. When isomerisation steps are required, the yield is predicted to drop strongly, with very-difficult-to-fold proteins that rely highly on isomerase activity showing only a fraction of the predicted yield of an otherwise equivalent easy-to-fold protein. Interestingly, the capacity to process isomerase-requiring substrates differs more strongly between conditions than non-isomerase-requiring ones, and the relationship is not proportional: for example, during growth in glucose the capacity to process an easy-to-fold substrate is predicted to less than during growth in glycerol, but the capacity to process a difficult-to-fold substrate is 4–5 times higher during growth in glucose. We assume that these differences reflect different proportions of DsbAB vs DsbCDG concentrations, and indeed examinations of the proteome datasets reveals that whereas concentrations of most Dsb enzymes are comparable under both growth conditions, the concentration of DsbC is increased during growth in glucose.
{kind=link}