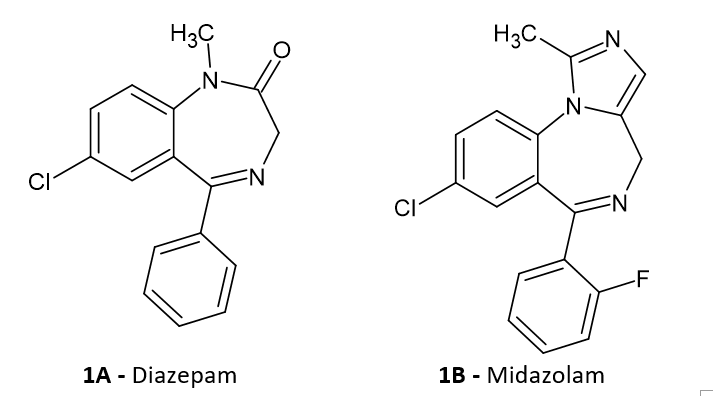
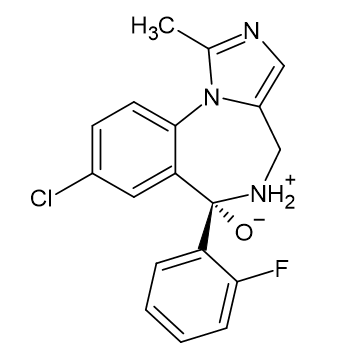
Midazolam
In the text the energetic data for B3LYP/6-311++G(d,p) results, both for the gas-phase and water environment modelled by PCM model are presented. The structure of midazolam (MDZ) optimized at the B3LYP/6‑311++G(d,p)/PCM level of theory is shown in Figure 1. All further Figures are done at this model chemistry.
From comparison of Scheme 1B and Figure 1 it follows that the molecule is in reality highly nonplanar. The methylene group is mostly responsible for this, it also breaks the pi-electron conjugation of the 7-membered ring and reduces significantly its aromaticity. It formally contains 7 pi-electrons (2 from the pyrrole-like nitrogen atom, common to both rings), which according to the Huckel rule makes it formally non-aromatic. The fused benzene ring as well as one connected by a single, rotatable bond are of course aromatic and planar, and so is the imidazole ring. To better understand the aromaticity of midazolam, two aromaticity indices were employed – geometric HOMA and electronic pEDA. However, calculating pEDA makes sense only for planar rings, thus for the benzodiazepine ring only HOMA is available. We will analyse here only 7- and 5-membered rings and will call accordingly HOMA for 7-membered ring - HOMA7, and HOMA for 5-membered ring - HOMA5. The pEDA index will be calculated only for the 5-membered ring and will be called - pEDA5. For midazolam HOMA7 = -0.30 and HOMA5 = 0.79, also pEDA5 = -0.07. It follows that the 7-membered ring is non-aromatic (negative HOMA) and 5-membered ring is aromatic with slight pi-deficiency (below the perfect electronic sextet). Later in the text the changes of HOMA and pEDA also for cations will be analysed.
Formation of carbinolamine
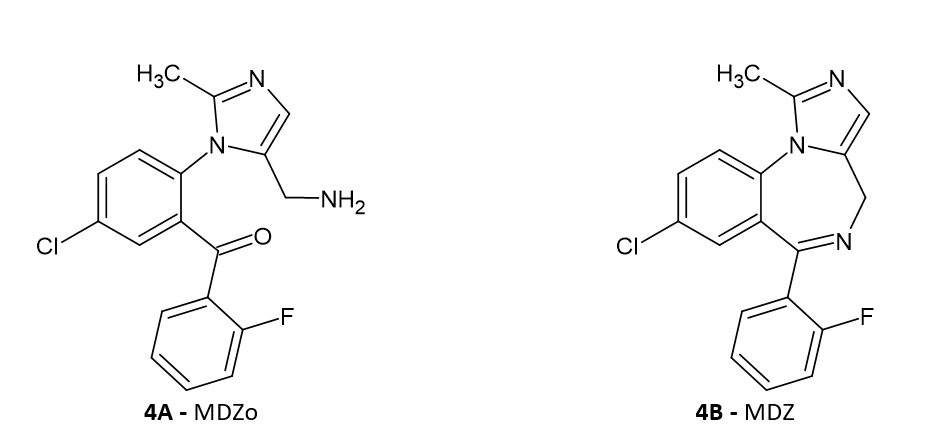
The first and essential step is the formation of carbinolamine via a transition state. Looking at the open form of midazolam it is virtually impossible to propose an initial structure of the transition state, sufficiently close to the true transition state, to obtain it by means of direct optimization. The most reliable procedure in such situations is to perform a relaxed potential energy surface scan. A series of geometry optimizations is performed, but in each case a chosen interatomic distance is kept fixed. In this case we found that the best procedure is to start not from the MDZo, but from carbinolamine. The atoms in consideration are the hydrogen from OH group and the nitrogen atom. We start from their distance in carbinolamine (2.6Å ) and gradually bring the hydrogen closer to the nitrogen atom, up to the distance of about 1.0 Å. The energy at first increases and when the structure is near the transition state, the energy is highest. Then, suddenly the ring opens (the proton now belong to the amino group), the energy lowers and the structure becomes the open ring form - MDZo. The structure of the highest energy from the scan is a good starting point for full optimization of the transition state. The substrate - MDZo – in proper conformation for upcoming nucleophilic attack, the transition state and the product – carbinolamine are depicted in Figure 2 below. All molecules are oriented spatially in the way to best show the essential bond lengths during the process of cyclization.
It follows from Figure 2 that the bond lengths in the transition state 2b is much closer to the product 2c that to the reactant 2a which is in accordance with the highly endoergic character of this reaction. The transition state much more resembles the product than the substrate which is in agreement with the Hammond’s postulate. The energetic parameters of the reaction are summarized in Table 1. Only the relative energetic parameters are given here, the absolute values are available as Supplementary Information.
Table 1. Relative values of total electronic energy, enthalpy and Gibbs free energy of molecules involved in carbinolamine formation, optimized at B3LYP/6-311++G(d,p) in gas-phase and water environment (PCM) for the first step of ring closure of midazolam
|
|
Gas-phase
|
Water environment (PCM)
|
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
|
2a
|
0
|
0
|
0
|
0
|
0
|
0
|
|
2b (TS)
|
33.84
|
31.38
|
35.08
|
32.64
|
30.22
|
33.93
|
|
2c
|
5.83
|
6.19
|
9.63
|
6.18
|
6.54
|
9.98
|
Regarding the thermodynamics, it follows from Table 1 that the first step of ring closure is an endothermic and endoergic process. The Gibbs free energy is even more positive than enthalpy because of the entropy loss during the process of ring closure – the number of degrees of freedom of the molecule is substantially reduced. With the carbinolamine less stable by almost 10 kcal/mol, it follows that it is created in very small amounts – the reaction equilibrium constant for this reaction is very low. In respect to kinetics, the calculated activation barrier is high enough – about 35 kcal/mol which results in limited speed of this reaction. Interestingly, the difference between gas-phase conditions and water environment is not large. In the later, the activation barrier is slightly reduced, but the product is slightly less stable thermodynamically.
The protonation of the carbinolamine and the water molecule loss
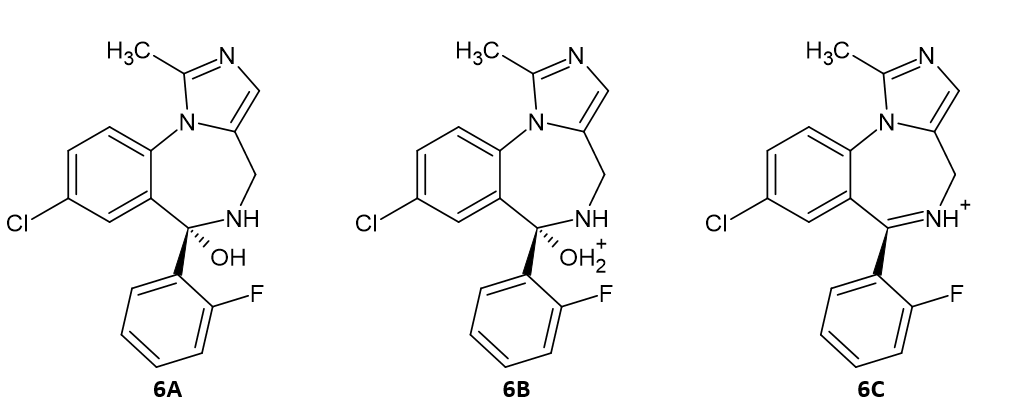
The next essential step in the formation of final midazolam molecule is the protonation of the hydroxy group of carbinolamine to facilitate the removal of the water molecule. In the idealized textbook image this is 6B transition product (Scheme 6). However, all attempts to obtain an energetic minimum of such a structure failed, because the water molecules immediately is expelled from the protonated carbinolamine molecule. We tried to perform a relaxed PES scan gradually approaching the H3O+ ion to the carbinolamine but the energy during the entire scan lowers and at certain distance the proton suddenly jumps to carbinolamine with immediately expelling the water molecule with further lowering of the energy. This shows that the process of protonation of carbinolamine is activation barrierless and there is no transition state involved. This reaction would undergo much faster in acidic environments, but as we must remember, the first step – the formation of carbinolamine demanded neutral of basic conditions. However, even in such conditions some hydronic ions exist in the water environment, so we can expect that protonation of carbinolamine occurs also in these conditions to some degree, which should be sufficient, because after protonation – it turns out – the protonated carbinolamine immediately breaks up into protonated imine and water molecule. Thus, it is not possible to obtain a stable transition product – protonated carbinolamine, except when we fix the length of the weak C-O bond at the value observed in neutral carbinolamine which is 1.450 Å for water environment (and 1.454 Å for the gas-phase). We decided to use the distance of 1.45 Å for fixed optimizations in both environments. The process of optimizing the geometrical parameters of a molecule is called constrained optimization. The result 3a is shown in Figure 3.
The geometry of structure 3a differs to some extend in regard to the structure 2c, for example the C‑N bond in the benzodiazepine ring is shortened from 1.44 to 1.42 Å. But in general, their geometrical shapes are very similar, and we can conclude that the protonation does not change much the geometry of the carbinolamine. If the C-O bond length is not fixed, there undergoes immediately the loss of water molecule from the protonated carbinolamine which results in protonated imine and water molecule complex 3b in Figure 3. It is needless to say that the process of expelling the water molecule is activation barrierless and there is no transition state. Comparison of structures 3a and 3b shows that the C-N bond now becomes clearly double, with its length equal to 1.3 Å. The relative energetic data for 3a and 3b are shown in Table 2.
Table 2. Relative values of total electronic energy, enthalpy and Gibbs free energy of protonated carbinolamine with fixed C-N bond length, optimized at B3LYP/6-311++G(d,p) in gas-phase and water environment (PCM)
|
|
Gas-phase
|
Water environment (PCM)
|
|
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
|
3a
|
0
|
0
|
0
|
0
|
0
|
0
|
|
3b
|
-58.97
|
-59.22
|
-62.15
|
-48.95
|
-49.80
|
-52.90
|
In water environment the enthalpy of 3b is lower by 50 kcal/mol and the Gibbs free energy by 53 kcal/mol than 3a. In gas-phase these values are even more negative by almost 10 kcal/mol. This is an illustration of great instability of 3a (if the C-O bond is not fixed). This reaction is connected with the gain of entropy, thus the more negative Gibbs free energy than enthalpy.
Acid-base equilibrium between two possible sites of protonation of MDZ
After the formation of the protonated imine, the rest is actually the question of acid-base equilibrium of MDZ molecule. As was mentioned in the introduction, there exist two possible sites of protonation – the benzodiazepine ring nitrogen atom and the imidazole ring nitrogen atom. The first possibility was already shown in Figure 3b. In the Figure 4 there are shown two possible tautomers of protonated MDZ (without external water molecule)
The Gibbs free energy difference between 4a and 4b is 1.1 kcal/mol for Gas Phase and 3.4 kcal/mol for water environment in favour of 4b at the B3LYP/6-311++G(d,p) level of theory, thus it is expected that the initially created form 4a will quickly tautomerize to more stable form 4b, especially in water environment. Let us look at the aromaticity of 5- and 7-membered rings for 4a and 4b, the data is gathered in Table 3.
Table 3. HOMA for 7- and 5-membered rings and pEDA for 5-membered ring for midazolam (MDZ) protonated at 7- and 5-membered ring.
|
HOMA7
|
HOMA5
|
pEDA5
|
|
4a
|
-0.098
|
0.774
|
-0.100
|
|
4b
|
-0.362
|
0.811
|
0.031
|
It follows from Table 3 that in midazolam cation protonated at 7-membered ring the aromaticity of this ring increases from HOMA7 = -0.30 to -0.10 but aromaticity of 5-membered ring is slightly reduced from HOMA5 = 0.79 to 0.77. Also the pi-deficiency of the 5-membered ring is slightly increased from pEDA5 = ‑0.07 to -0.1. On the contrary, in case of protonation at the 5-membered ring HOMA7 = -0.36 so it is slightly reduced, comparing to midazolam, but HOMA5 = 0.81 which means slight increase. Also pEDA5 is now positive – the ring is little pi-excessive. These changes are all rather small. We can look at it also from a more visual perspective – the pattern of single and double bonds shows a better bond conjugation and equalization of the 5-membered ring in the case of MDZ protonated at imidazole (Supplementary Information - Figure S1). Similar picture of better bond equalization cannot be seen in case of protonation of 7-membered ring. Possible sources of this include: (1) the methylene group which breaks the conjugation of bonds and (2) the fused benzene ring interferes with bond equalization. Thus, the protonation at the imidazole nitrogen atom is much more beneficial.
Deprotonation of the protonated imine to MDZ
The final step in our chain of reactions is the deprotonation of the protonated imine to actual closed-ring midazolam MDZ. This process should be easiest of course in basic conditions where the OH- ions are available. To estimate if the whole chain of reactions is endothermic or exothermic we have to guarantee that the number of atoms and electron is exactly the same for the molecules which we compare. Otherwise, any comparisons of the energy of two molecules have no physical sense. Fortunately we can achieve this by comparing the MDZo with MDZ complex with a water molecule. In this case there is exactly the same number of atoms and electrons as the whole reaction is just an amine-carbonyl condensation. Also the charge of the molecules is the same – both are neutral. Thus, we need the MDZ molecule with a water molecule attached to it. There are two possible comparisons: of water molecule attachment: (1) to benzodiazepine ring nitrogen atom and (2) to imidazole ring nitrogen atom. The first possibility is a “natural” one as we can expect that when on OH- ion is close to 3b, the proton will be removed and the water will be attached to the seven-membered ring. However, because of the possible tautomeric equilibrium described in the previous chapter and for the sake of completeness, we should also make the comparison with MDZ with water molecule attached to imidazole ring.
The Gibbs free energy difference between 5a and 5b is 1.3 kcal/mol for gas-phase and 1.9 kcal/mol for water environment at the B3LYP/6-311++G(d,p) level of theory and the more stable molecule is again 5b. Lower energy of 5b is also reflected in stronger hydrogen bond between water molecule and MDZ, a rough measure if this strength is the bond length, which is a little shorter in case of 5b (see Figure 5). The comparison of energetic parameters of the first molecule – midazolam open (benzophenone) and final molecule – midazolam closed (+ water molecule) shows the thermodynamics of the whole chain of reactions. The data is summarized in Table 4.
Table 4. Relative values of total electronic energy, enthalpy and Gibbs free energy of closed form midazolam with water molecule attached to nitrogen atom of 7-membered and 5-membered ring in respect to open form, optimized at B3LYP/6-311++G(d,p) in gas-phase and in water environment (PCM)
|
|
Gas-phase
|
Water environment (PCM)
|
|
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
|
MDZo
|
0
|
0
|
0
|
0
|
0
|
0
|
|
5a
|
-4.66
|
-5.12
|
-5.64
|
-7.18
|
-7.72
|
-7.67
|
|
5b
|
-5.96
|
-6.42
|
-6.94
|
-7.91
|
-8.55
|
-9.53
|
It follows that the whole chain of reactions is exothermic and exoergic, with similar enthalpy and Gibbs free energy difference, no matter to which final product we make the comparison. There is one subtle difference – in water environment if we make the comparison to 5a, there is neither entropy gain or loss because the loss in the formation of carbinolamine was compensated by the gain in the process of water secretion. In the case of comparison to 5b the is about 1 kcal/mol lower than . In the gas-phase the Gibs free energy for both comparisons is lower by about 0.5 kcal/mol. We can conclude that from the thermodynamic point of view the reaction with progress completely to the closed form of midazolam – the reaction equilibrium constant for the whole chain of reactions is high.
From the kinetic point of view the reaction rate limiting stage is the first step – the formation of carbinolamine with its quite high activation barrier – over 30 kcal/mol. The rest of the process undergoes fast without activation barriers.
The model of midazolam
In order to understand what is the impact of other fragments of the molecule on the thermodynamics and kinetics of benzodiazepine ring closure, we constructed a model where all other parts of the molecule except the imidazole ring were removed and replaced by hydrogen atoms. The model is shown in Figure 6 and will be called MMDZ.
Regarding the aromaticity of the model, the indices are: HOMA7 = -0.07, HOMA5 = 0.84 and pEDA5 = -0.005. It follows that both rings are more aromatic than in midazolam itself. Thus, it follows that other parts of the molecule decrease the aromaticity of the core rings.
Formation of carbinolamine
The first step was to model the carbinolamine molecules formation for which reactant, transition state and product are shown in Figure 7.
From a quick comparison of Figures 7 and 2 it follows that there are significant differences in geometries of MDZo and MMDZo and transition states. The carbinolamines show a very similar geometry. Regarding the open forms the C-N distance is much smaller in the model (2.9 Å comparing to 3.3 Å). It is probably connected to simpler structure of the model with less steric repulsions, allowing for more flexibility of the chain of atoms in the model. In the transition state all essential bond lengths (except C-N) are longer in the model. The most interesting is however the energetic data which is gathered in Table 5.
Table 5. Relative values of total electronic energy. enthalpy and Gibbs free energy of molecules involved in carbinolamine formation, optimized at B3LYP/6-311++G(d,p) in the gas-phase and water environment (PCM) for midazolam model
|
|
Gas-phase
|
Water environment (PCM)
|
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
|
7a
|
0
|
0
|
0
|
0
|
0
|
0
|
|
7b (TS)
|
32.44
|
30.27
|
33.26
|
40.48
|
38.62
|
41.59
|
|
7c
|
-6.15
|
-4.91
|
-2.20
|
-5.48
|
-4.35
|
-1.83
|
When comparing Table 4 to Table 1, the most striking fact is that the process of carbinolamine formation is exothermic and exoergic in the case of the model of midazolam. A possible explanation of this fact is that for midazolam, the closure of the ring is connected with many unfavourable steric interactions. The open form of midazolam has very complicated spatial structure, its conformation is heavily bent in several directions to allow for the energy lowering. During the ring closure many bonds have to rotate and the closed carbinolamine puts many constrains on the structure. On the other hand, the structure of the model is much simpler and its closure does not involve these unfavourable interactions.
The protonation of carbinolamine and the water molecule loss
The protonated carbinolamine with C-O bond length fixed at 1.446 (the value for optimized model carbinolamine) is unstable, like in the case of MDZ. The important difference is that in the case of model it was not possible to obtain a true minimum, but only the first order saddle point. It follows from comparison of Figures 8 and 3 that the C-O and C-N bonds are shorter in the model. Without constrains the structure 8a immediately loses the water molecule during optimization leading to 8b.
The energetic data for the water loss process are gathered in Table 6.
Table 6. Relative values of total electronic energy, enthalpy and Gibbs free energy of protonated carbinolamine with fixed C-N bond length, optimized at B3LYP/6-311++G(d,p) in the gas-phase and in water environment (PCM)
|
|
Gas-phase
|
Water environment (PCM)
|
|
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
∆H
[kcal/mol]
|
∆E
[kcal/mol]
|
∆G
[kcal/mol]
|
|
8a
|
0
|
0
|
0
|
0
|
0
|
0
|
|
8b
|
-50.03
|
-50.28
|
-54.38
|
-38.96
|
-39.65
|
-44.06
|
The difference between 8b and 8a is smaller than in the case of 3b and 3a by about 10 kcal/mol. It can be explained by the nature of 8a which is a first-order saddle point, thus is richer in energy than a minimum.
Acid-base equilibrium between two possible sites of protonation of MMDZ
As in the case of MDZ there exist two possible tautomeric forms of MMDZ cation and they are showed in Figure 9.
It turns out however that the order of stability of these form is reversed. The molecule protonated at the 7-membered ring nitrogen atom - 9a has lower Gibbs free energy. In gas-phase the difference is 1.1 kcal/mol and in water environment it is increased up to 2.8 kcal/mol. In order to better understand these data let us look at the aromaticity indices gathered in Table 3.
Table 3. HOMA for 7- and 5-membered rings and pEDA for 5-membered ring of midazolam model protonated at 7- and 5-membered ring.
|
HOMA7
|
HOMA5
|
pEDA5
|
|
9a
|
0.242
|
0.645
|
-0.296
|
|
9b
|
-0.201
|
0.841
|
-0.005
|
Comparing the situation of protonation at 7-membered ring and 5-membered ring we see that aromaticity of 7-membered ring significantly increases in the first case and significantly reduces in second case. For the 5-membered ring the situation is reversed, but the changes of aromaticity are much smaller. The pEDA5 = -0.30 for 9a shows some pi-electron deficiency of the 5-membered ring and means that the pi-electron density is shifted to 7-membered ring. If we look at the bond equalization scheme (Figure S2 – Supplementary Information) we can see a conjugated, butadiene-like fragment in the 7-membered ring in case of 9a. The fused benzene ring is not present in the model thus it does not interfere with the bond equalization pattern and this increases the aromaticity of the 7-membered ring, thus stabilizes the cation protonated at the 7-membered ring. This ring is less non-planar than analogous ring in the midazolam cation.
Deprotonation of the protonated imine to MDZ
Finally, let us compare the open-ring form of midazolam model to its closed form with a water molecules attached, again to guarantee the equal number of atoms for energy comparisons. The neutral form MMDZ with a water molecule attached are showed in Figure 10.
The energetic situation becomes now very interesting as both molecules 10a and 10b are very close in energy. In gas-phase the 10a has slightly lower energy and in water environment 10b but the difference is less that 0.5 kcal/mol so it is below the 1 kcal/mol error and is not meaningful. We can assume that 10a and 10b have very similar stability. It follows from Figure 10 that the hydrogen bond length (connected to its strength) is only a little shorter for 10a. On the contrary in Figure 5 this bond for 5b is much shorter than 5a which is connected with better stability of 5b. Thus, a comparison of the final product – midazolam model with water molecule to midazolam model open will be done only for 10a.
Table 7. Relative values of total electronic energy, enthalpy and Gibbs free energy of closed form midazolam with water molecule attached to nitrogen atom of 7-membered ring in respect to open form, optimized at B3LYP/6-311++G(d,p) in the gas-phase and in water environment (PCM)
|
|
Gas-phase
|
Water environment (PCM)
|
|
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
∆E
[kcal/mol]
|
∆H
[kcal/mol]
|
∆G
[kcal/mol]
|
|
MMDZo
|
0
|
0
|
0
|
0
|
0
|
0
|
|
10a
|
-8.67
|
-8.96
|
-10.54
|
-9.88
|
-10.33
|
-11.53
|
It follows from Table 7 that the whole ring closure process for the model is even more exothermic and exoergic than for the midazolam itself. It can be attributed to less congested structure of the model of the midazolam for which the process of ring closure not connected to any unfavourable steric interactions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}