Differentiation to IPC can be achieved from various cellular sources such as: embryonic stem cells (ESC) [28], induced pluripotent stem cells (iPSCs) [29] and MSC derived from adult tissue, as adipose (hASC) [13] [14], bone marrow (BM-MSC), [30] dental pulp (DPSCs) [31], and placenta [31]. Use of ESC and iPSCs in clinic is not routinely approved due to ethics and implementation considerations. Yet, hASC can be easily obtained from low invasive lipoaspiration procedures with high yield of cell purification. Hence, these cells constitute an important source of MSC [11, 12].
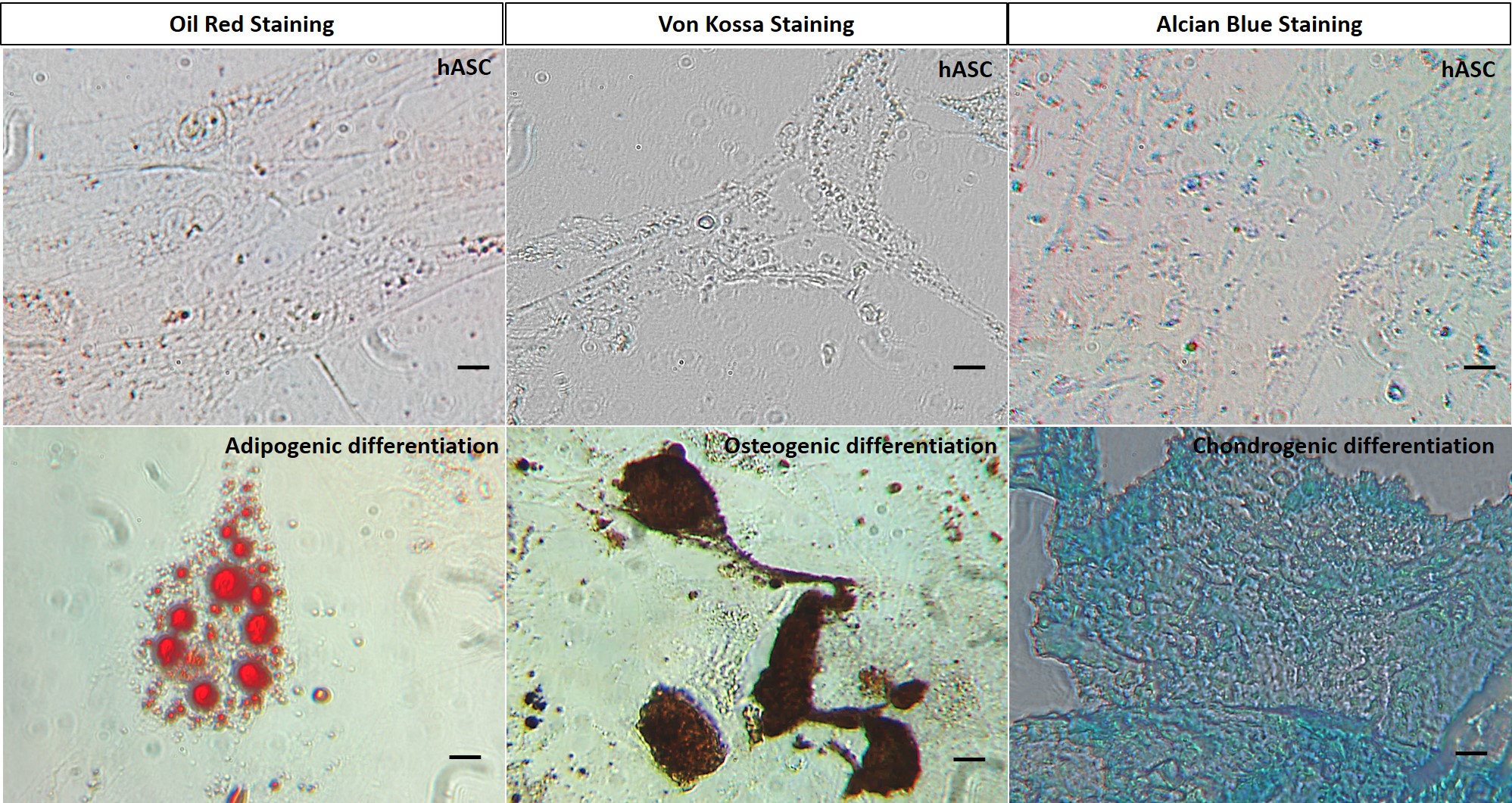
The hASC obtained in this work meet the requirements dictated by the International Society of Cell Therapy (ISCT) to classify a cellular population as a MSC [27, 32, 33]. Our cells were able to adhere to plastic with a fibroblastic phenotype, they expressed CD90, CD73, CD105, CD44, and CD29 while lacking expression of CD45, CD34, CD19 and HLA-DR, concordant to the surface marker expression described for hASC [11, 12]. Moreover, they were able to differentiate to adipogenic, chondrogenic and osteogenic phenotypes.
We then established a reliable method to induce differentiation of this cells into both IPC and GPC in vitro. Our results agree with those of other groups, in particular the differentiation process of hASC to IPC, [13, 14]. On the other hand, differentiation to GPC have only been reported from ESC, [15]. To the best of our knowledge, we are the first group to achieve an efficient and functional GPC differentiation from hASC using the protocol described by Rezania group, yet using SFB 2% instead of the B27 supplement. In line with these findings, we propose hASC as an adequate cellular source to use in cell based therapy against T1D.
Next, we co-cultured IPC and GPC in a 4:1 ratio by inducing cellular aggregation in low adhesion conditions, as previously described [26]. Using this setting, we observed that cell aggregates increased in size up to 72 h, reaching diameters of ~ 80 µm, similar to those reported for human islets (108 µm) [34]. Expression of α and β pancreatic cell markers in 72 h old cellular aggregates was also conserved, indicating a strong lineage commitment and low dedifferentiation rate of our cells.
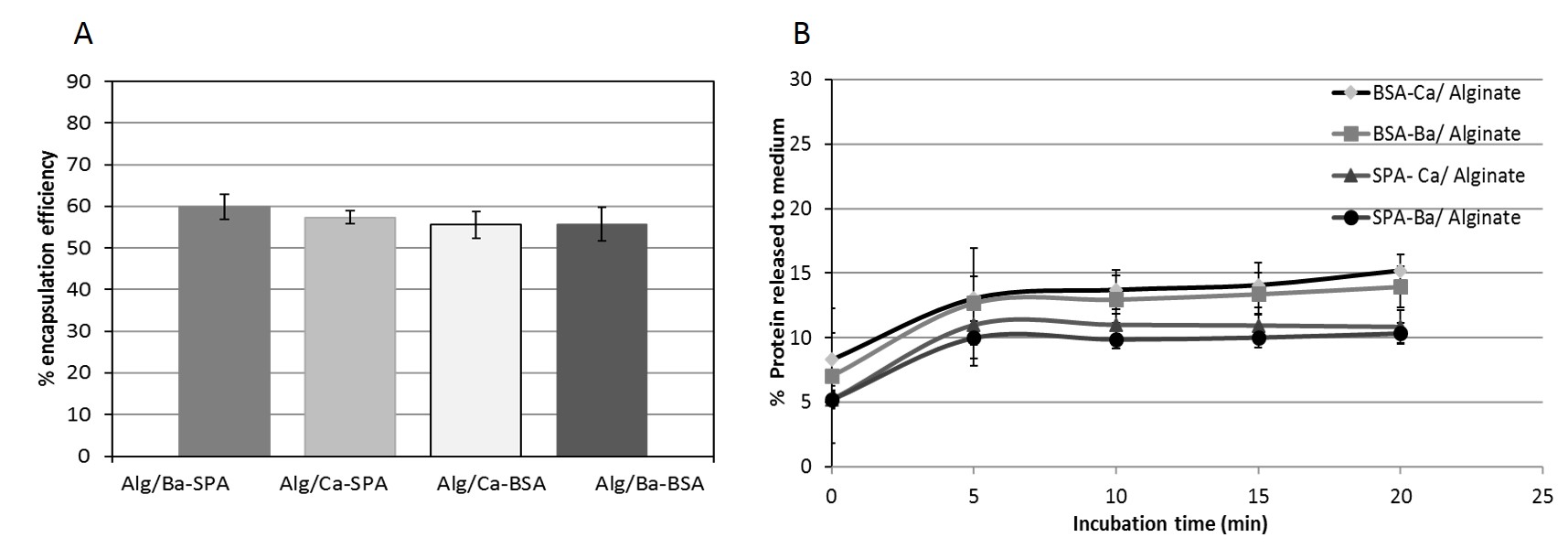
Although human like islets should be able to partially restore the pancreatic endocrine function, the activation of the immune system is still a major limitation, together with the low number of donors for long term treatment [6, 8]. To overcome this issue, we encapsulated our IPC/GPC aggregates into sodium alginate microgels using an automatized encapsulator. The size of Ca2+ or Ba2+ stabilized sodium alginate microgels pools were very similar, with very low dispersion. In culture conditions, Ca2+ stabilized microgels increased in size and were less stable then Ba2+ stabilized microgels, which maintained their size and shape even after 7 days of culture. An explanation to this phenomenon lies in the chemical characteristics of the sodium alginate copolymer, which is composed by 1,4-b-D-mannuronic acid (M) and 1,4-a-L-guluronic acid (G) residues, and can be stabilized in general terms by divalent cations being Ba2+ the most efficient (Mg2+ << Ca2+ < Sr2+ < Ba2+) [19].
Several studies have aimed to improve the output of encapsulated tissue grafts using sodium alginate. However, variables such as alginate purity and immunological response against residual contaminants left from the purification process are still a matter of discussion, [19]. In our setting, both Ca2+ or Ba2+ stabilized alginate microgels were able to release both BSA and SpA proteins in the first 5 minutes, yet after 20 minutes the release rate diminished and remained stable. This information defined the alginate microgels pore size, which in essence permits the passage of nutrient such as glucose, as well as small peptides as Ins and Ggc. In agreement with these findings, we also confirmed that our microencapsulated cellular aggregates responded to external glucose by secreting Ins, thus confirming that the microgels allowed the passage of glucose into the microgel to be sensed by the cellular aggregates, which in turn responded by secreting insulin.
Despite the stability of the microgels, IPC/GPC aggregates ceased to secrete insulin after 7 days in culture. This may be attributable to loss of β cell function due to a dedifferentiation process, a desensitization of the IPC to glucose, or poor survival of the cellular aggregate in encapsulation conditions in the in vitro environment. Indeed, the described differentiation protocols attempted to reproduce the embryogenic process observed in organs and tissues in vitro. Yet the complexity of the embryogenic process in cell differentiation differs substantially from the much simpler in vitro milieu, a situation that could result in dedifferentiation, and in turn loss of phenotypic and functional characteristics. Nevertheless, an in vivo environment may overcome such limitations. We are currently planning experiments with in vivo implantation of our encapsulated clusters in NOD mice, an animal model of T1DM, which may shed light on this issue.
Our findings enforce the current knowledge on the potential of hASC as a source of multipotent cells in cellular therapy against T1DM. Moreover, we have developed a protocol to generate functional aggregates IPC/GPC like pancreatic islets, which can be encapsulated in alginate microgels to achieve immunoprotection. In such conditions, the encapsulated aggregates are stable and retain endocrine function in vitro for at least 7 days in culture.
{kind=link}
{kind=link}