Environmental factors shaped the skin microbiota community structure and diversity
To profile the skin microbiota of healthy cattle, skin scurf was sampled from 25 different herds located in northeast to east Australia (Fig. 1 and Table S1) and the 16S rRNA gene amplicons were sequenced from microbial DNA isolated from each sample. The skin microbial community structure was visualised using a Bray-Curtis similarity-based multidimensional scaling (MDS) plot (Fig. 2A). Each cattle herd appeared to have distinct microbial community structures, with herds 19 and 20 being the most divergent to the rest of the herds. These two herds were located at the northernmost sampling site within a proximity of 3 km to each other (Fig. 1).
Associations between environmental variables and microbiota community ordination were examined to determine the factors driving the observed differences in the skin microbiota. This identified significant correlations of the microbial community structure with the location of the farm (altitude, latitude, and longitude) and specific climatic parameters (solar exposure, temperature, and rainfall) (Fig. 2A). For example, altitude and latitude were found to be the strongest parameters driving the microbiota differences in herds 22 and 24, whilst rainfall and temperature (seven-day average prior to sample collection) contributed significantly to shaping the skin microbial community in herd 8. Upon examination of the impact of host factors such as the breed, we observed no significant correlation with the microbiota community structure, suggesting that differences in the microbiota community structure between herds were mainly due to environmental variables.
The microbiota alpha diversity, as determined using a Shannon diversity index, was generally high across most herds, indicating a diverse skin microbiota in healthy cattle (Fig. 2B), this is consistent with previous reports [1]. Herds 3, 19 and 20 had lower alpha diversity indices compared to all other samples. To examine whether this is due to an impact of environmental variables, we conducted pair-wise correlation analyses between Shannon diversity indices and geographic and climatic information. This revealed a significant negative association (Pearson correlation = − 0.43, P < 0.05) between the Shannon diversity indices and latitude of the farms. Other tested environmental variables had no significant association with microbiota diversity. While our results suggest an impact of the location, particularly latitude, on the diversity of cattle skin microbiome, future studies with sampling sites spanning a broader geographical area are essential for confirming this observation.
Some Bacterial Phyla And Families Were Highly Abundant Across All Cattle Herds
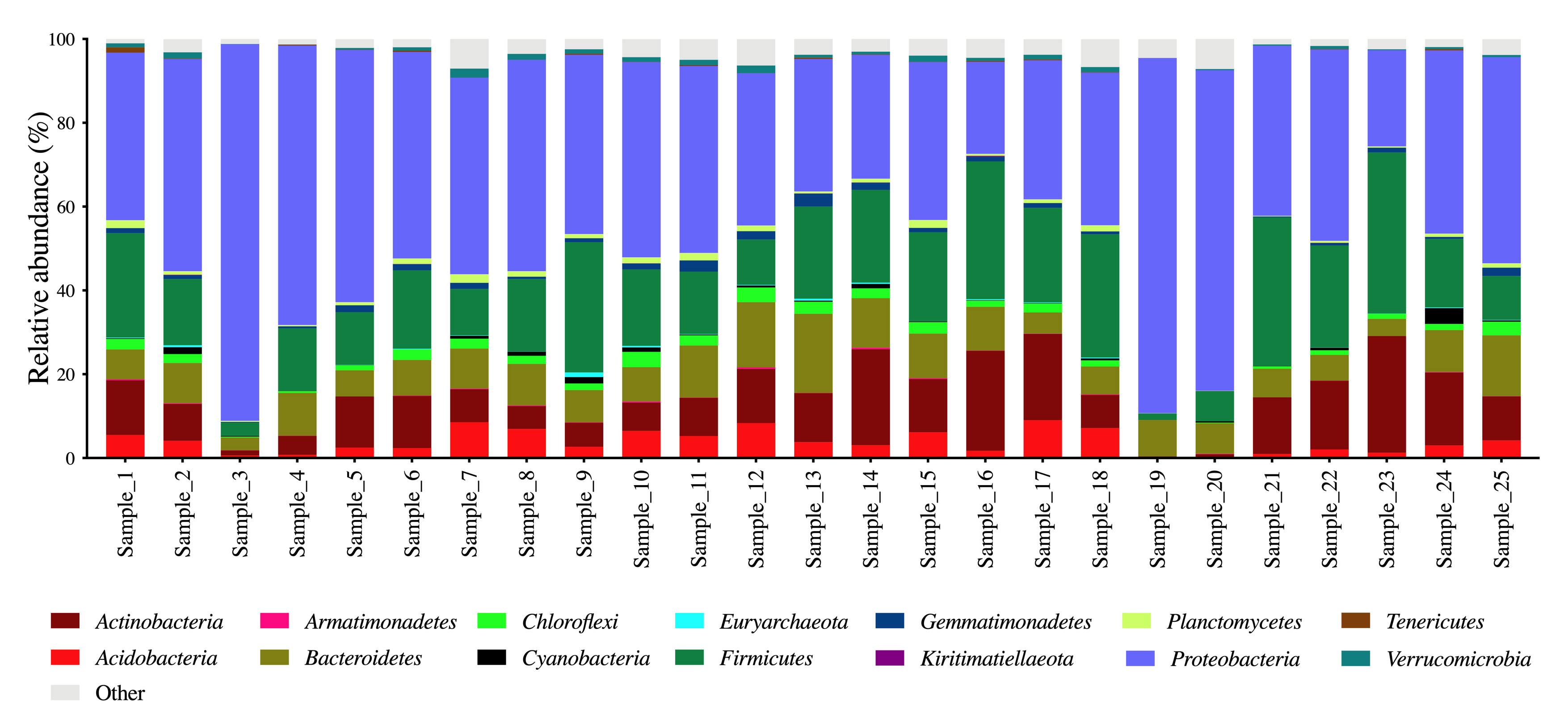
Despite the differences in microbiota community structure and diversity, some microbes were consistently identified on the skin of most cattle. For example, upon examination of the relative microbial abundance at the phylum, we identified 35 different bacterial phylum assignments across all skin microbiota samples. Among these, the phyla Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes and Acidobacteria were highly abundant in the skin microbiota of all herds (Figure S1). Microbial abundance at the order and family level was assigned to 210 orders and 378 families (Table S2A), demonstrating a complex and diverse community in the skin microbiota of healthy cattle. Several bacterial families including Moraxellaceae, Sphingomonadaceae, Bacillaceae, Burkholderiaceae and Peptostreptococcaceae were observed on the skin of all 25 herds (Fig. 3 and Table S2B). A total of 7,476 amplicon sequence variants (ASVs) were identified across all 25 cattle herds (Table S3). Of these, 234 ASVs, some of which were in the families Moraxellaceae, Sphingomonadaceae and Bacillaceae, were highly abundant across most herds.
While similar types of bacterial phyla and families were seen in most samples, their relative abundance significantly differed between herds. Notably, a higher abundance of Proteobacteria and a lower abundance of Actinobacteria and Acidobacteria were seen in cattle with a lower microbiota alpha diversity (herds 3, 19 and 20) compared to the herds with a higher microbiota alpha diversity (Fig. 2B and Figure S1). Similar variations in the microbiota composition were observed at the family level (Fig. 3). For example, in herds 3, 4, 19 and 20, more than 50% of the taxa were assigned to the family Moraxellaceae, a member of the phylum Proteobacteria that is commonly observed in both human and animal skin [5] including the cattle skin microbiota [18].
Identifying core microbes that are present on the skin irrespective of the host and environmental variables is vital for developing microbiota-based strategies to manage skin disease and pest infestation in cattle. Our study is the first to investigate this using 25 herds of Australian cattle. The most abundant bacterial phyla (Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes) which were observed across all herds (Figure S1), have been previously reported in healthy bovine digits and teat skin [1, 19, 20]. Moraxellaceae, Sphingomonadaceae, Bacillaceae, Burkholderiaceae and Peptostreptococcaceae are bacterial families generally present on the teat and digit skin of healthy cattle [18, 21–24]. Despite these similarities, investigations on the cattle skin microbiota thus far have been limited to specific body sites. Our study addresses this knowledge gap through profiling the microbial communities living in the skin of cattle.
Location And Climate Had Significant Impacts On The Abundance Of Skin Bacteria
To examine the impact of environmental factors on the observed variations in the relative abundance of skin microbial communities, pair-wise correlation analyses were conducted. These revealed significant associations between specific environmental parameters and the abundance of specific bacterial families (Table 1). Solar exposure and rainfall significantly contributed to the differences in microbiota composition across herds. For example, the relative abundance of Bacillaceae, Planococcaceae, Staphylococcaceae and Micrococcaceae positively correlated with solar exposure. The first three families, which are all in the class Bacilli, contain members that are resistant to desiccation [25–27]. The family Micrococcaceae, a member of the phylum Actinobacteria, is tolerant to heat [28]. The higher abundance of these families in the skin microbiome of cattle located in areas with a higher solar exposure is likely explained by their ability to tolerate physical challenges including desiccation. The abundance of the family Sphingomonadaceae was found to be higher in areas with greater rainfall (Table 1). In agreement with our observations, members of this family have been previously reported in freshwater [29], a potential source introducing this bacterial family onto the cattle skin.
Table 1
Pearson’s correlation coefficient of pair-wise correlation analysis between the relative abundance of bacterial families and environmental variables.*
| Bacterial Family | Latitude | Longitude | Altitude | Rainfall | Solar exposure |
| Bacillaceae | | | | | + 0.40 |
| Staphylococcaceae | | | | | + 0.48 |
| Micrococcaceae | | | | | + 0.41 |
| Planococcaceae | | | | | + 0.50 |
| Sphingomonadaceae | + 0.45 | + 0.52 | | + 0.44 | |
| Cellvibrionaceae | + 0.45 | + 0.42 | | | |
| Chitinophagaceae | + 0.44 | | | | |
| Pseudomonadaceae | + 0.43 | + 0.45 | | | |
| Rhodopirillaceae | + 0.43 | | | | |
| Weeksellaceae | | | + 0.47 | | |
| *Coordinates of statistically significant (p < 0.05) correlations are shown. + denotes a positive correlation. |
The location of the farm (latitude, longitude, and altitude) showed significant positive correlations with the abundance of six bacterial families: Cellvibrionaceae, Chitinophagaceae, Pseudomonadaceae, Rhodosprillaceae, Sphingomonadaceae and Weeksellaceae. However, there were no significant associations between these families (except Sphingomonadaceae) and any of the tested climatic parameters (temperature, rainfall, or solar exposure). Members of the Cellvibrionaceae, Chitinophagaceae and Sphingomonadaceae have been found in soil and water [30, 31], suggesting that observed associations could be related to environmental or host factors not tested in this current study.
The current study examined the overall skin microbiota of cattle (along the backline and flanks). Future investigations on whether microbes on different skin sites respond differently to climatic and environmental changes will be useful for comprehensively understanding the commensal skin microbiota of cattle. The herds in this study were in northeast and east Australia and their skin scurf was sampled once. Sampling herds from a larger geographical area and at multiple times throughout the year will contribute to extending the outcomes of the current study and provide a thorough understanding of the commensal skin microbiota of cattle and how seasonal changes influence it.
Alterations in the skin microbiota have been associated with various skin diseases and inflammatory conditions in both humans and animals [4, 24, 32]. Skin health in cattle is paramount for the wellbeing of animals and profitability in the dairy and beef industries. While there is an increase in the recognition of the role of the skin microbiota in regulating cattle skin health, there is a lack of studies directly examining this in livestock. Elucidating how external factors that impact the skin microbiota of healthy cattle will not only be critical for developing novel microbiome therapies but will also facilitate designing better studies to examine the microbiota alterations in various skin diseases.
{kind=link}