Agm-fitc And Protein Microarray
Agm-FITC has been described in previous studies [16]. Briefly, Agm (0.16 mg, Sigma-Aldrich) was added to a DMF (1 ml, Fisher) solution containing FITC (50 mg, Thermo) and TEA (18 ul, Sigma-Aldrich) at room temperature (RT). The organic solution was washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified using column chromatography (CH2Cl2: MeOH = 10:1) to obtain a product with a 60% yield. To screen the target proteins that bind to Agm, we used the HuProt human proteome microarray v4.0 (CDI Laboratories). This chip was blocked with 20 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.1% Tween-20, and 5% BSA on an orbital shaker for 2 h, at RT. For screening Agm binding proteins, the blocked chip was washed 3 times with a microarray buffer for 10 min, followed by incubation with Agm-FITC (2 ug/ml) for 8 h at 4°C. After washing each chip three times with 1x PBS-T and Milli-Q water, the microarray was centrifuged for 5 min in a 50 mL centrifuge tube and scanned with an Axon GenePix 4000B Microarray Scanner (Molecular Devices). The probe signals of the microarray were acquired using the GenePix Pro6.0 software (Molecular Devices). After scanning, the probes were considered detectable when the z-scores for both duplicates were over three. The mean of the signal intensities from all proteins on the chip was calculated.
Cell Culture And Drug Treatment
The murine microglia cell-line (BV2) was obtained from the Ajou University College of Medicine, Chronic Inflammatory Disease Research Center. BV2 was cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Thermo) with 10% fetal bovine serum (FBS; Thermo) and 1% penicillin/streptomycin (Thermo) and maintained in a 37°C environment containing 5% CO2. To induce the M1 phenotype activated by lipopolysaccharides (LPS) or the M2 phenotype activated by IL-4, BV2 was treated with LPS (20 ng/ml, Sigma-Aldrich) and IL-4 (20 ng/ml, cell signaling), respectively. The cells were also simultaneously treated with Agm (100uM, Sigma-Aldrich). Cultured media containing the BV2 cells were transferred to serum-free RPMI-1640 media after 24 h of stimulation, where they were cultured for a further 48 h.
Co-immunoprecipitation (Co-ip)
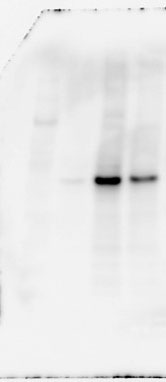
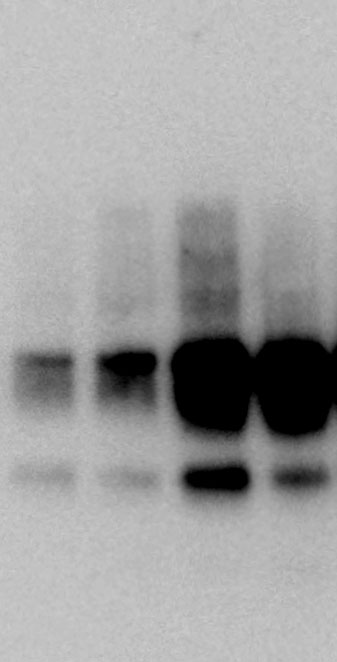
BV2 lysates were precleared using Protein A/G Agarose (50 ml, Santa Cruz). The rabbit anti-IRF2BP2 (2.5 mg, Thermo), rabbit anti-Interferon regulator factor 2 (IRF2; 2.5 mg, Abcam), and mouse anti-IgG isotype control (2.5 mg, Santa Cruz) were added to the precleared lysates (200 ml) and incubated overnight at 4°C. The Protein A/G Agarose was collected and the supernatant was aspirated by micro-centrifugation (3,000 g for 2 min at 4°C). This procedure was repeated twice. After washing all the reactions three times, the products were boiled for 5 min and thereafter micro-centrifuged briefly to pellet Protein A/G Agarose.
Nucleus And Cytoplasm Fractionation
To isolate the nucleus and cytoplasm of BV2, a NE-PER Nuclear Cytoplasmic Extraction Reagent kit (Thermo) was used according to the manufacturer’s instructions. Briefly, The BV2 pellet was centrifuged at 500 g for 3 min and thereafter suspended in 200 µl of cytoplasmic extraction reagent (CER) I by vertexing. Following this, 11 µl of CRE II was added to the suspension and centrifuged at 16000 g for 5 min. The cytoplasmic extract was transferred to a pre-chilled tube, to which 100 µl of the nuclear extract was added, and centrifuged at 16000 g for 10 min. The resulting supernatant, constituting of both nuclear and cytoplasm extracts, was used for further experiments.
Immunoblotting (Ibt)
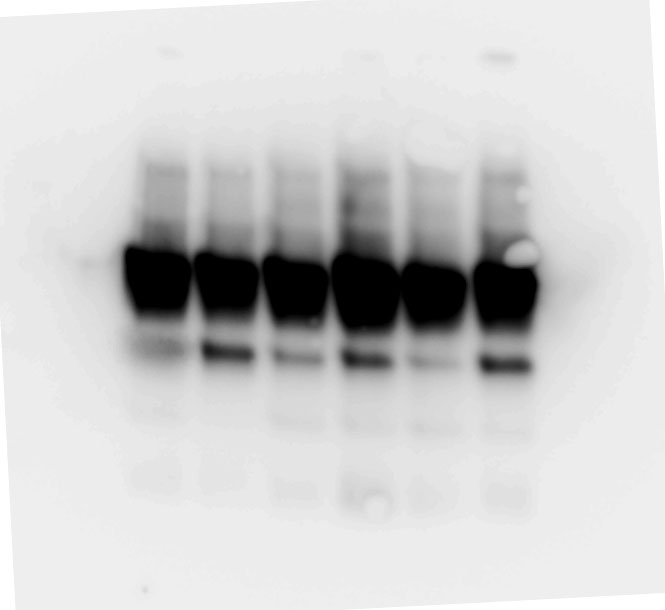
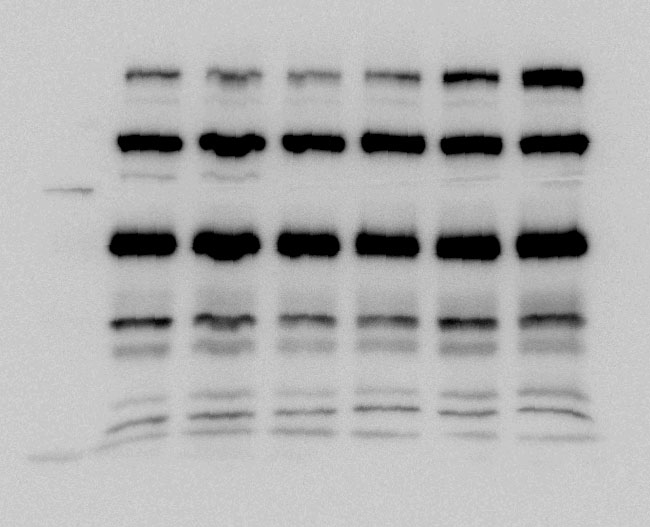
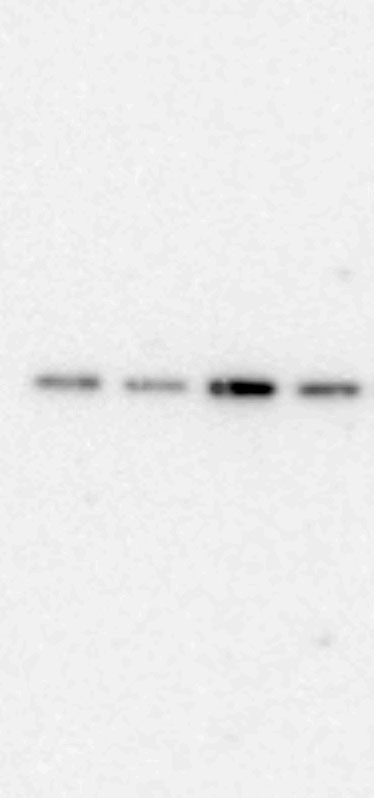
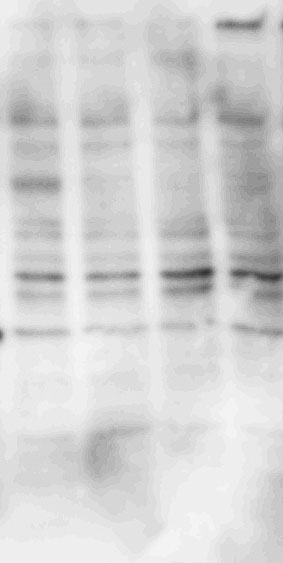
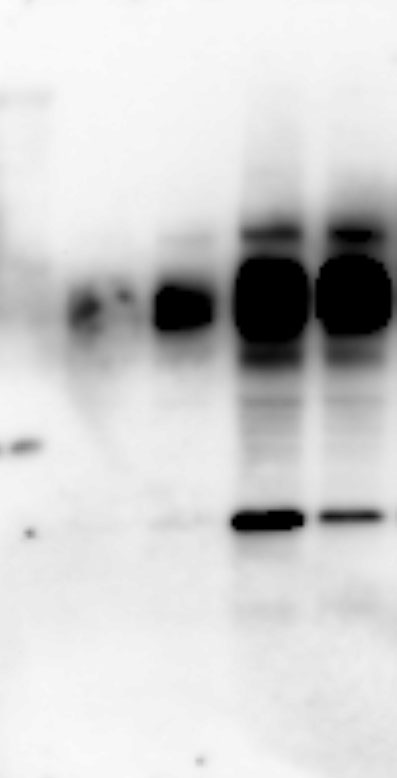
Whole proteins were isolated using ice-cold RIPA lysis buffer (Tech & Innovation) containing 1 mM PMSF (Thermo). The whole protein (50 ug), Co-ip sample (50 ug), and nuclear and cytoplasm extract (30 ug) were separated on 10% sodium dodecyl sulfate-polyacrylamide electrophoresis gels (Bio-Rad), and thereafter electro-transferred onto a polyvinylidene difluoride membrane (Milipore). The intensity of the proteins was probed with rabbit anti-IRF2BP2 (1:1000, Thermo), rabbit anti-IRF2 (1:1000, Abcam), mouse anti-INF-ꝩ (1:1000, Abcam), mouse anti-KLF4 (1:1000, R&D system), mouse anti-CD86 (1:1000, Abcam), rabbit anti-CD206 (1:1000, Abcam), goat-anti-Lamin B (Santa Curz), and rabbit anti-β-actin (1:1000, Abcam) overnight at 4°C. The membrane was reacted with goat anti-Mouse IgG(H + L)-Peroxidase conjugated HRP, goat anti-rabbit IgG(H + L)-Peroxidase conjugated HRP, and rabbit anti-goat IgG(H + L)-Peroxidase conjugated HRP for 1 h at RT. Immunoreactive bands were detected using the ECL system (Thermo) and visualized using the LAS-4000 (Fuji).
Immunocytochemistry (Icc)
BV2 were fixed using 4% PFA for 15 min and permeabilized using 0.1% Triton X-100 for 7 min. All the cell groups were washed three times for 3 min at each step with PBS. Before treating them with the antibodies, the cells were blocked using 3% BSA for 1 h. Rabbit anti-IRF2BP2 (1:1000, Thermo), rabbit anti-IRF2 (1:1000, Abcam), mouse anti-KLF4 (1:1000R&D, system), mouse anti-CD86 (1:1000, Abcam), and rabbit anti-CD206 (1:1000, Abcam) were incubated overnight at 4°C. After washing the cells three times with PBS, we treated them with donkey anti-Rabbit IgG Alexa flour 488 (1:1000, Invitrogen,) and donkey goat IgG Alexa flour 568 (1:1000, Abcam). Finally, the cells were stained with the mounting solution DAPI (Thermo) and visualized using the LSM 700 (Carl Zeiss).
Electrophoretic Mobility Shift (Emsa) Assay
To confirm the interaction between IRF2 and Kruppel-like factor 4 (KLF4) promoter, an EMSA kit (Thermo) was used according to the manufacturer’s instructions. DNA probes of KLF4 (5′-GGT AGT GGG GAA TGG GAA AAG GAG T-3′, IDT) were also prepared. Briefly, the nuclear extract was obtained according to the nuclear fractionation method mentioned above. Biotin-labeled DNA-binding oligonucleotides were incubated with 10 mg of nuclear extract at 15°C for 30 min, to allow the formation of the KLF complex. The DNA binding complexes were separated from the free probes using a 6% polyacrylamide gel. Following electrophoresis, the gel was transferred to a nylon membrane at 380 mA for 45 min. DNA crosslinking was performed. Once the blocking process was complete, the membrane was washed with washing buffer three times at RT. Signals were observed using enhanced chemiluminescence reagents. Images were captured using the LAS-4000 (Fuji).
Flow Cytometry
To analyze BV2 polarization by Agm, the cells (5 x 106/ml) were washed with FACS buffer (PBS with 0.5% BSA) and blocked using 3% BSA. The cells from each sample were treated with Mouse anti-CD86 labeled with APC (BD, 1:1000) and Mouse anti-CD206 labeled with PE-Cy7 (BD, 1:1000), for 1 h. The negative controls were treated with compensation beads negative control (BD, 1 drop) and compensation beads anti-mouse Ig,k bead (BD, 1 drop). The positive control samples were only treated with compensation beads anti-mouse Ig,k bead (BD, 1 drop); thereafter the APC or PE-Cy7 control antibody (BD,1:1000) was added to each sample. Finally, all the samples were analyzed by following BD FACS LSR II SORP (BD).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}