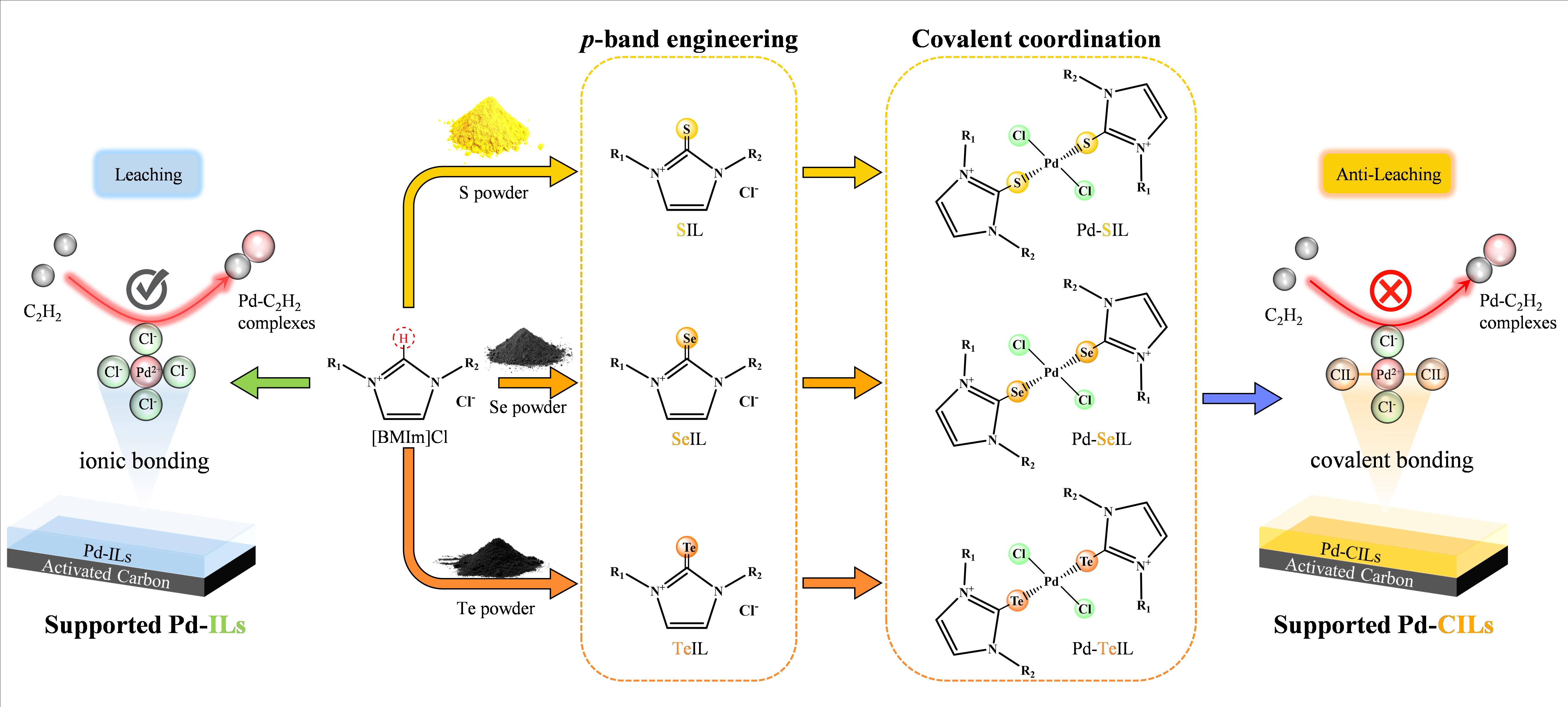
Synthesis and structural identification of Pd-CIL/AC catalysts.
Initially, we synthesized three kinds of CIL covalent ligands, named [BMImS]Cl, [BMImSe]Cl and [BMImSe]Cl, respectively, by a simple thermochemical method using 1-butyl-3-methylimidazolium chloride ionic liquid and elemental sulfur, selenium or tellurium33. Supplementary Fig. S1 is 1-butyl-3-methylimidazolium thione ionic liquid ([BMImS]Cl), yellow liquid. Supplementary Fig. S2 is 1-butyl-3-methylimidazoliumselenone ionic liquid ([BMImSe]Cl), light brown liquid. Supplementary Fig. S3 is 1-butyl-3-methylimidazolyl tellurone ionic liquid ([BMImTe]Cl), light red liquid. In order to verify the structure of the covalent ligand, the corresponding 1H-NMR characterization analysis of the ligand was carried out. The NMR characterization results were consistent with those reported by Inesi et al29. The infrared results also prove that there are corresponding C = S, C = Se and C = Te functional groups in the three covalent ligands, respectively (Supplementary Fig. S4). Therefore, this facile thermochemical synthesis method can be used for the facile and rapid synthesis of target CIL covalent ligands.
ILs-containing Pd-based catalysts (Pd-IL/AC and Pd-CIL/AC) were synthesized by a conventional wet-impregnation method, employing an aqueous solution of the corresponding precursors14,22−24. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) revealed small Pd clusters in the unfunctionalized IL sample (Fig. 1a), which disappeared after introduce CIL implying their dispersion into single atoms. The sole presence of isolated palladium atoms is confirmed by aberration-corrected HAADF-STEM (AC HAADF-STEM) (Fig. 1b-d) and extended X-ray absorption fine structure spectroscopy (Fig. 2a), indicating a strong interaction between CIL and Pd. In contrast, the absence of ionic liquids resulted in severe sintering of impregnated Pd species into sub-micron Pd NPs aggregates (Supplementary Fig. 5), consistent with our observations for Au sintering over AC following the wet impregnation. The dispersion of the atoms may be characterized through quantitative analysis of the atom positions. Histograms of the nearest neighbor distances measured for the investigated ILs-containing catalysts are shown in Fig. 1e-h. Based on a statistical analysis of above 300 Pd sites, Pd-IL/AC (Fig. 1e) shows an average nearest-neighbour distance of 1.23 nm. While regions of well-dispersed single atoms were identified in the sample, AC HAADF-STEM imaging evidenced some metal dimers with atom-atom spacings (< 0.3 nm) significantly smaller than the mean nearest neighbor distance. The ratio of Pd interatomic distances less than 0.3 nm is about 8.9% in Pd-IL/AC, which means that there are a few of Pd clusters/nanoparticles in the system, since the nearest neighbor atomic distance of Pd in Pd bulk phase is 0.279 nm. Conversely, Pd-SIL/AC (Fig. 1f) shows an average nearest-neighbour distance of 1.64 nm. At such separation distances there is no coupling between atoms. Nearly no Pd-Pd distance within 0.3 nm can be observed, excluding the possibility of any Pd clusters or nanoparticles in our catalyst. Similar results are seen for the Pd-SeIL/AC and Pd-TeIL/AC catalysts in Fig. 1g and Fig. 1h, respectively. Since the Pd/AC and Pd-IL/AC samples transitions through lower temperatures during the preparing process, we reasoned that these PdClx species must be thermodynamically unstable and hence should be susceptible to leaching when subject to a further high-temperature reaction. TG analysis confirmed that acetylene treatment of the PdCl2 powder sample resulted in complete loss of the Pd component (Supplementary Fig. 6). Representatively, the spatial uniformity of individual elements in the Pd-SIL/AC is confirmed in the elemental maps acquired by energy-dispersive X-ray spectroscopy (Fig. 1i).
To identify the pivotal structural parameters of Pd-CIL/AC for the synthesis of high-loading atomically dispersed Pd catalysts, extended X-ray absorption fine structure (EXAFS) spectroscopy was recorded, as EXAFS spectroscopy is very sensitive to the local environment of metal atoms35–38. Accordingly, EXAFS was applied to confirm the presence of single metal atom sites in the various Pd-CIL/AC. Figure 2a shows Pd K-edge EXAFS data (R space plots) for the SACs along with data for relevant reference samples. For each Pd-CIL/AC, the R space plots were quite distinct from that of the corresponding Pd foil reference (Supplementary Fig. 7). Pd-SIL/AC showed an intense peak at approximately 1.45 Å, corresponding to the first Pd-Cl/S coordination shell, with the oscillation being very similar to that observed for PdS and PdCl2, indicating that Pd atoms in the Pd-SIL/AC had a similar local environment with that of Pd atoms in PdS/PdCl2. Fitting result determined that Pd in Pd-SIL/AC was coordinated fourfold by Cl/S/ClxSy (x + y = 4) atoms, as is also found in Pd-SeIL/AC and Pd-TeIL/AC (Fig. 2a). The Pd-SeIL/AC and Pd-TeIL/AC also showed only a single peak in R space (at shorter lengths than the typical Pd-Pd distance in the corresponding Pd foil), confirming that no Pd-Pd bonds existed in the samples. These results were in good accord with the findings of the HAADF-STEM analyses. Note that, the interatomic distances follow an order of Pd-TeIL/AC > Pd-SeIL/AC > Pd-SIL/AC, as shown in Fig. 2b. The first coordination shell scattering distance in the Pd-CIL/AC increases with increasing chalcogen atomic number and is differential to the interatomic distance for Pd-Cl. These results suggest the presence of a well-defined Pd-S/Se/Te phase. The coordination numbers (CNs) of Pd in the Pd-SIL/AC catalyst is ca. 4.0, and the CNs of Pd-Cl and Pd-S are both about 2 (Supplementary Table S1), suggesting that the Pd-S coordination in Pd-SIL/AC catalyst was successfully constructed. Considering that the sulfur species are mainly in the form of -C = S, we therefore presume that, in these atomically dispersed catalysts, two -C = S and two Cl− moieties coordinated with one Pd atom to form the Pd-S2-Cl2 mononuclear complex-like structure (Fig. 2f), which typically endowed the heterogeneous Pd species with homogeneous catalyst properties. Similar single-atom Pd-Se2-Cl2 and Pd-Te2-Cl2 structures were obtained in the Pd-SeIL/AC and Pd-TeIL/AC catalysts (Fig. 2c & 2f, Supplementary Table S1).
To further strengthen this result, the wavelet transforms (WT) analysis of Pd EXAFS oscillations was conducted (Fig. 2d and Supplementary Fig. 8), giving powerful resolution in both k and R spaces. As illustrated by the overlay WT contour plots of Pd foil, PdCl2 and PdS (dark, green, and red regions in Fig. 2d), the intensity maxima at ~ 12 Å−1, ~ 8 Å−1, and ~ 3 Å−1are attributed to the Pd-Pd, and Pd-Cl, and Pd-S contributions, respectively. In contrast, for the WT contour plot of Pd-CIL/AC (yellow for Pd-SIL/AC, navy blue for Pd-SeIL/AC, and blue for Pd-TeIL/AC; Fig. 2d), intensities maximum between ~ 3 Å−1 and 8 Å−1 are exclusively observed, which are assigned to the Pd-S-Cl, Pd-Se-Cl, and Pd-Te-Cl contribution, respectively. A linear combination fitting (LCF) analysis of the Pd K-edge XANES spectra was employed to quantitatively assess the Pd coordination composition in Pd-CIL/AC, using XANES spectra of PdCl2, PdS, PdSe, and PdTe standards. The results of the analysis for the Pd-CIL/AC are shown in Fig. 2e. The comparison of the spectra and the LCF results show similar trends for each Pd-CIL/AC, with Pd-S/Se/Te and Pd-Cl components accounted for approximately 50%. The best-fitting analyses clearly confirm that Pd center is coordinated with two Cl atom and two chalcogen atoms, as illustrated in Fig. 2f. According to the calculated configuration energy of Pd-CIL constructures (Supplementary Fig. 9), trans configuration has lower energy than the cis configuration, therefore, trans configuration is thermodynamically favorable. Therefore, the results of AC-HAADF-STEM, XPS and XAFS characterizations verify the formation of trans Pd-CIL2-Cl2 with atomically dispersed Pd atoms on the surface of AC.
Electronic properties.
To analyze the interactions, we have calculated the electron localization function (ELF) and localized orbital locator (LOL), which can effectively reveal the degree of electron localization and the bonding character between individual atoms39–42. The bonding motif is nicely illustrated by the ELF and LOL maps in Fig. 3a-e (Supplementary Fig. 10), which shows the electron–density accumulation of the Pd-Cl ionic-bonds and of the in-plane Pd–S/Se/Te covalent-type bond, as well as the multiple maxima of the lone-pair electrons. In details, ELF shows that in the Pd-IL liquid sample, there is no obvious electronic localization feature between Pd and Cl, indicating that the interaction between the two is mainly ionic bond. However, in PdS configuration, the electron density is highly localized in the internuclear Pd–S bonding region, indicating that Pd-S is distinct covalent bond. One can also see pronounced maxima of the ELF near S atoms, corresponding to the lone electron pair of the sulfur atom. Similarly, The ELF distribution in Pd-SIL shows strong charge transfer from S to Pd, indicating that the free electron gas density of Pd-S is quite high, and forms large amount of Pd–S covalent bonds (Fig. 3c). In view of the excellent thermal stability of PdS, we speculate that the construction of Pd-S bonds can greatly improve the durability of Pd-SIL during the reaction process. Compared with the ELF of Pd-S bonds (0.87) for Pd-SIL, the incorporation of Se and Te will induce a lower ELF of 0.85 and 0.79 for the Pd-Se and Pd-Te bonds in Pd-SeIL and Pd-TeIL, respectively (Fig. 3d&e), which indicates Pd–Se and Pd-Te bonds are more ionic than Pd-S bonds. Hence, the Pd-S bonds in Pd-SIL furnishes higher chemical bond strength than Pd-Se and Pd-Te in Pd-SeIL and Pd-TeIL, implying that Pd-SIL is more thermodynamically stable. Meanwhile, the calculated partial density of states (PDOS) shows that the sharing range of Pd and Cl in Pd-IL is very small, while both S 2d and Pd 3d of Pd-SIL have the substantial DOS in the sharing range from − 4 to 2 eV, suggesting the strong electronic coupling between Pd and S atoms (Fig. 3f). The strong electronic coupling between Pd and Se/Te atoms was also observed in Pd-SeIL and Pd-TeIL. Interestingly, it was found that with the functionalization of ionic liquids, the sharing range of Pd and Cl became larger, which promoted the interaction between Pd and Cl. In addition, compared with the Pd d-center for Pd-IL, the Pd d-centers for Pd-CIL can be observed with an obvious downshift relative to Fermi level after the construction of Pd-CIL sites. As a result, the electronic structure of Pd atoms for Pd-CIL could be effectively regulated with more bonding states which would enable the active sites to have a weaker binding ability with reactive intermediates, thus inhibiting the generation of Pd-C2H2 complex.
XANES spectra and k3-weighted K-space spectra for the different Pd-CIL are shown in Fig. 3g. For all three Pd-CIL samples, the adsorption edge was higher than that of the corresponding Pd foil, indicating that Pd-CIL contained Pd atoms in cationic states (likely the Pd2+ state). The chemical composition and elemental states of the Pd-CIL/AC were investigated by XPS. The Pd 3d XPS spectrum for Pd-CIL (Fig. 3h) showed a Pd 3p5/2 peak at 337.1 ~ 337.4 eV, higher than that observed for Pd0 (335.1 eV) and slightly lower than that for Pd2+ in Pd/AC. The data provide strong evidence for the presence of Pd2+ in the Pd-CIL, with the slightly lower binding energy compared to Pd/AC due to coordination by chalcogen atoms rather than Cl. The C 1s spectrum (Supplementary Fig. 11a) was deconvoluted into two peaks, corresponding to C-C or C = C (neutral carbon and adventitious hydrocarbons) and C-N species15,43−46. The N 1s spectrum (Supplementary Fig. 11b) for the Pd-CIL could be deconvoluted into one peak, corresponding to C-N = C species45–47. The S 2ps spectrum (Supplementary Fig. 12a) was deconvoluted into two peaks, corresponding to C-S and and MSx species (where M stands for metals)48–49. The presence of MSx, which originated from the SIL, adds further weight to the proposal that the Pd atoms in the Pd-SIL/AC existed in Pd-S coordination environments (as established from the EXAFS analyses). The XPS data collected for the other Pd-CILs (Supplementary Figs. 11–12) were similar to that of Pd-SIL, with the metal core-level spectra providing strong evidence for the presence of Pd-Se and Pd-Te coordination.
Reactivity, durability, and recyclability of Pd-CIL/AC catalysts.
First, we evaluated the catalytic performance of the catalysts at different reaction temperatures and GHSV(C2H2), as is shown in Fig. 4a-c, the color in two-dimensional maps represents the YVCM of catalysts. In comparison, it is found that all Pd-CIL/AC catalysts have nearly 100% YVCM at low temperature (< 120°C) and low GHSV(C2H2) (< 200 h− 1), and also show more than 70% YVCM at high temperature (> 160°C) and high GHSV(C2H2) (> 2000 h− 1). Interestingly, Pd-TeIL/AC and Pd-SeIL/AC exhibited higher YVCM at low temperature (< 120°C) and high GHSV(C2H2) (> 500 h− 1), however, the catalytic performance in the high temperature region (> 150°C) was slightly lower than that of Pd-SIL/AC catalyst, indicating that the catalytic performance of Pd-SIL/AC catalyst is better in the high temperature range. To identify a speciation-sensitive activity descriptor, kinetic studies were performed over the whole Pd-CIL/AC catalyst platform. A correlation between initial activity and increasing apparent activation energy (Ea) can be identified (Fig. 4a), with the exception of Pd/AC catalysts. Each of these three samples is expected to have a slightly lower affinity towards both substrates, implying the reduced activation, slowing the surface catalytic reaction rates. The overall reaction orders with respect to HCl and C2H2 were derived for Pd/AC, Pd-IL/AC and Pd-CIL/AC and were found to be in the range 0.5–1.1, indicating that the reaction rate depends on the surface coverage of both reactants (Supplementary Fig. 13). In most cases, a higher partial reaction order is obtained for HCl (nHCl ≈ 0.7–0.8) than for acetylene (nC2H2 ≈ 1.0–1.1), suggesting the former has a more critical role in the reaction mechanism, which contrasts with a previously reported result. These results hint at the possible key role of substrate affinity in balancing the interaction with the reactants and the durability of the catalyst. Since stability is critical for practical application of catalysts for acetylene hydrochlorination, the long-term evaluation over the prepared catalysts were investigated, the results are shown in Fig. 4e. During 2000 h reaction, a slightly decrease in YVCM from 99–96% was observed for Pd-SIL/AC, which was apparently higher than that of other catalysts14–17, thereby demonstrating its excellent long-term stability. In the 1200 h reaction, the Pd-IL/AC catalyst lost its activity drastically. However, compared to Pd-IL/AC, Pd-SeIL/AC and Pd-TeIL/AC showed better hydrochlorination activity; the YVCM of Pd-SeIL/AC and Pd-TeIL/AC decreased from 99–97% and 99–96%, respectively, after 500 h of continuous testing. We observed above that both catalysts have relatively high low-temperature activity than Pd-SIL/AC, indicating a slightly higher intrinsic activity than that of Pd-SIL/AC catalyst, which may lead to higher thermal effects to initiate side reactions, eventually leading to catalyst deactivation. We therefore use the Pd-SIL active phase as a representative to study its stability and recyclability in the next experiments.
To demonstrate the generality of the strategy, investigations on catalyst deactivation behavior were conducted while loading Pd-IL and Pd-SIL active phases over different porous supports, including oxide support, carbon nitride, and molecular sieves. After 48 h reaction at 180°C and GHSV(C2H2) 740 h− 1, we noted that all supported Pd-SIL phase catalysts exhibit lower deactivation rate (< 0.4% h− 1) than those of supported Pd-IL catalysts on the same carrier (Fig. 4f). Immediately afterwards, the Pd content of the used catalysts were measured by ICP-MS and compared with the fresh catalysts. For AC carrier, all supported Pd-CIL catalysts show a leaching percentage below 0.15 wt.% after 48 h reaction, which is drastic lower than that of Pd/AC (64.7 wt.%) and Pd-IL/AC (13.2 wt.%). In addition, we separately tested the Pd weight loss after reaction over investigated supports. After 48 h reaction, as is shown in Fig. 4g and Supplementary Table 2, ICP-MS analysis of the used catalyst indicted < 0.4 wt.% loss in all catalysts compared with the corresponding fresh catalysts, especially the loss of Pd on activated carbon fiber (ACF) was almost negligible (< 0.02 wt.%).
Encouraged by the extraordinary durability of Pd-SIL active phase during reaction process, we carried out a series of additional thermal treatment experiments at temperatures ranging from 120 to 280°C under acetylene atmosphere. Experimentally, we find that the Pd/AC catalyst showed serious leaching of Pd active components in acetylene atmosphere and increased exponentially with the increase of heat treatment temperature. On the other hand, with the increase of the treatment temperature, the loss rate of Pd has been maintained at about 0.3%, while the loss of fresh catalyst measured by the same method is 0.37%, which is mainly due to the insufficient catalyst digestion and loss of solution during transfer process. The recyclability of metals in catalysts is also one of the most important properties for practical applications1,50−53. In this process, the Soxhlet extraction method was firstly used to extract the Pd phase in the catalyst and then the Pd concentration was determined. Multiple Soxhlet extraction experiments were conducted to study the recyclability of the Pd-SIL/AC catalyst. First, by using acetonitrile as extractant, the fresh Pd-SIL/AC catalyst was extracted at 120 C for 72 h, and the process needed to keep the cooling water refluxing at all times to prevent acetonitrile from escaping. Then, a certain amount of the extract was taken out and diluted to a certain multiple and the concentration of Pd in it was measured by ICP. The remaining dilution liquid was poured back into the mother liquor and reloaded onto fresh activated carbon to determine its reusability. This extraction, detection and reloading step is repeated ten times. The catalytic activity of Pd-SIL/AC before and after extraction are shown in Fig. 4i, and the right ordinate represents the concentration of Pd in the solution after each extraction. After ten cycles, the recovered catalyst retained almost constant catalytic activity, and the concentration of Pd in the extract also fluctuated around 0.025 mg ml− 1, which was almost consistent with the theoretical value, as indicated by the ICP-MS analysis (Fig. 4i). All the above experimental results well explain the excellent stability and recyclability of the Pd-SIL/AC catalyst.
In order to verify the universality of the strategy, anti-leaching experiments were also conducted for Hg and Ru catalysts, which are prone to loss during the reaction process. It was found that SIL was also able to form covalent bonds with Hg and Ru species to improve the dispersion of active centers on the activated carbon surface and inhibit the loss of active centers, and both catalysts had good recyclability. The related details are presented in the Supplementary Figs. 14–15 and Supplementary Table 3.
We noted that a slow deactivation of the Pd-SIL/AC catalyst occurred during the reaction for up to 2000 hours. Therefore, to further investigation the stability of the catalyst and the deactivation mechanism, we carried out a comprehensive structural characterization analysis of the used catalyst. The combination of XRD, XAS, XPS, HAADF-TEM, BET, and elemental analysis are used to analyze the possible reasons for deactivation. Surprisingly, HAADF-TEM (Fig. 5a) and XRD (Fig. 5c) investigations verified the presence of only single copper atoms in the used Pd-SIL/AC catalyst, while the spatial uniformity of individual elements in the used Pd-SIL/AC is confirmed in the elemental maps acquired by energy-dispersive X-ray spectroscopy (Fig. 5b). There is no discernible difference between the XAS and XPS plots of the used catalysts (Fig. 5d-e, Supplementary Fig. 16) and those of the fresh catalysts, indicating that the coordination of Pd sites was maintained both before and after the reaction. These findings further demonstrate the high appropriateness of the SIL ligands to stabilize single Pd atoms, even in long-term reaction circumstances. As a result, we conclude that the deactivation of Pd-SIL/AC is not caused by changes in the active sites. Instead, there are differences in the porous properties and elemental composition of the fresh and used catalysts (Supplementary Figs. 17 and 18), with the used Pd-SIL/AC showing a slightly decrease in surface area and pore volume (887 to 755 m2 g− 1 and 0.49 to 0.40 cm3 g− 1, Supplementary Table 3), indicating the coke deposition (from 0.85 to 4.94 wt.%) during reaction process. These findings suggest that coking might lead to the pore blockage, which probably caused the catalyst deactivation for Pd-SIL/AC. Ultimately, these findings indicate that, unlike unfunctionalized ILs, functionalized ILs ligands provides appropriate bonding strength of entrapping single Pd atoms under reaction circumstances; however, it still suffers from slightly pore obstruction, which may result in catalyst deactivation.
Activity and durability descriptor
To identify a speciation-sensitive durability descriptor, DFT investigations were performed over the whole Pd-CIL catalyst platform. First, to unlock the bonding information of different CILs with Pd center, the crystal orbital Hamilton population (COHP) was conducted to evaluate its bonding strength54. Since more bonding states and fewer anti-bonding states yield stronger bonding and vice versa, the ICOHP analysis is directly connected to the binding energy results (Fig. 6a). For COHP, the integral area value represents the bonding strength of the hybrid covalent bond. The COHP results presented in Fig. 6a, indicate that a much larger proportion of occupied antibonding states near EF in Pd-IL, while the occupied antibonding states are significantly reduced when coordinated with CILs, especially the introduction of SIL makes COHP without any antibonding states below − 1.5 eV, demonstrating that SIL has unique anchoring site distribution and heterogeneous bonding strength compared to ILs. Furthermore, by computing the energy barrier that must be overcome for chemical bond breakdown, we may compare the stability of various coordination bonds intuitively. Findings shown in Supplementary Fig. 19, indicate that Pd-Cl in Pd-IL exhibits minimum energy barrier (0.22 eV) compared with that of Pd-S (2.15 eV), Pd-Se (2.04 eV), and Pd-Te (1.96 eV) in Pd-SIL, Pd-SeIL and Pd-TeIL, respectively, demonstrating that the covalent coordination significantly enhanced the interaction between CILs and Pd active sites.
Since CILs has been proved to have significant promoting effect on Pd catalysts stability (anti-leaching). As follows, we make an attempt to elucidate the unique stability in CILs system thorough theoretical computation. Electronic structure descriptors are computationally efficient quantities used to construct qualitative correlations for a variety of properties. In particular, the p-band center has been used to guide material discovery and fundamental understanding of an array of perovskite compounds for use in catalyzing the oxygen reduction and evolution reactions55–58. Here, we studied trends in catalytic performance as a function of Pd d-band center and the ligands (CILs and Cl) p-band center. Notably, the p-band center of ligands as a function of Pd leaching for all catalysts (Fig. 6b), in consistent with the deactivation rate, pinpointing its potential use as a stability descriptor. The ligands with lower p-band center exhibit higher anti-leaching properties and therefore showed lower deactivation rate. In line with the stability gained through the introduction of CIL ligands, identified by DFT, the stabilization is maximized on Pd-SIL/AC catalysts (Supplementary Fig. 19). Furthermore, we first evaluated the acetylene hydrochlorination performance using the differential adsorption energies, Eads(C2H2) – Eads(HCl) (ΔEads), as a unified descriptor accounting for the intrinsic thermodynamic stability (Fig. 6c). Notice that the adsorption energy of HCl on all IL-contained catalysts (Fig. 6c) is larger than C2H2, while the adsorption energy of C2H2 on PdCl2 (-0.86 eV) is much larger than that of HCl (-0.48 eV). In addition, the ΔEads values follow the order of Pd-SIL > Pd-SeIL > Pd-TeIL > Pd-IL > PdCl2, which is qualitatively consistent with the stability experiments obtained from the corresponding catalysts. It has been reported that the position of the coordinating atom (CA) p-band center, relative to the Fermi level, can be a descriptor and it is related to the hybridization of metal 3d and coordinating atom 2p orbitals. Here, we note that the charge transfer gap between CA 2p and metal 3d orbitals would be narrow with uplifting the CA p-band center, which can change the electron localization distribution, reaction kinetics and chemisorption properties. Accordingly, introduction of CILs ligand, and changes in ligand p-band center, shift the configuration towards the more stable conformer.
Having identified the relevant factors that govern the stability of Pd-CIL-based catalysts in acetylene hydrochlorination, we extended the scope of our investigation to activity descriptors. In stark contrast, the d-band center is virtually insensitive to activity. The correlation between metal d-band center of the catalysts with activity is generally the strongest, but no meaningful correlation can be observed between the experimental TOF and the d-band center of catalysts (Supplementary Fig. 20), disqualifying the d-band center as an activity descriptor. To identify a speciation-sensitive activity descriptor, thorough adsorption performance was performed over the whole Pd-CIL catalyst platform. Reaction mechanism analysis was further carried out to figure out the underlying factors governing the remarkable catalytic activity differences of the studied model catalysts. Figures 6d and 6f show the structural models of the paths of transformation for the conversion of acetylene to VCM on the investigated Pd active centers. The models are established based on EXAFS fitting results. The DFT calculation results show that the adsorption of C2H2 on the Pd-IL has better thermodynamic characteristics than those of Pd-CILs. The conversion of *C2H2 to *C2H2Cl is the stability-determining step. Due to the strong adsorption of Pd to C2H2, the C ≡ C bond connected to Pd is elongated, which promotes the breaking of the C ≡ C bond, making the free energy of this step lower. Meanwhile, due to the strong adsorption of Pd to C2H2, the coordination bonds of Pd with ligands was greatly weakened, which in turn leads to the leaching of Pd active components. In Pd-IL model, the reaction barrier of *C2H2 (Ads1) to *C2H2Cl (IM) is the lowest than that of Pd-CIL models, which is more likely to be converted to *Pd-C2H2, confirming the stability of Pd-CIL catalysts. In addition, the DFT calculation results (Supplementary Fig. 19) also show that bond breaking energy barrier for Pd-Cl (~ 0.2 eV) is the smallest, much lower than those of Pd-CIL (> 1.9 eV). Subsequently, *C2H2Cl (IM) will complete the activity-determining step of addition reaction to *CH2CHCl through the continuous action of *Pd-HCl. Because the H-Cl bond in Pd-IL is elongated the longest, the breaking of the H-Cl bond is easier than those in Pd-CIL. Thus Pd-IL (− 0.45 eV) has a lower free energy than Pd-CIL (> -0.6 eV), and Pd-IL is more likely to have the highest reactivity. To further confirm this hypothesis, we quantitatively compared the reaction activation energy (Ea) and reaction heat (ΔE) of investigated models (Fig. 6e). There is a linear relationship between the dissociative adsorption energy of HCl molecule (Ads2) and the activation barrier of HCl decomposition (TS2). In this step, the Ea and ΔE show a good linear relationship, which satisfies the typical Brønsted-Evans-Polanyi (BEP) relationship. Thus, a framework and hierarchy of property-performance relationships were identified to account not only for activity, but also stability (Fig. 6).
{kind=link}