A novel multi-responsive diarylethene derivative (1O) by linking a fluorescein group has been synthesized. Colorless 1O not only displayed good photochromic property to form cyclization state 1C, but also the 1C having multi-responsive characteristic to Cu2+ and basic stimulations. After Cu2+ was introduced, 1C showed excellent absorption and fluorescent spectra changed upon binding with Cu2+. The color of 1C solution turned from purple to colorless and the corresponding fluorescence color changed from dark to yellow, and the binding stoichiometry of 1C to Cu2+ was 1:1 and the detection limit was 0.11 µM. Moreover, 1C displayed distinctly reversible change on absorption and fluorescent spectra in basic environment, the basic environment can discolor the solution and turn on the fluorescence effect, suggesting that 1C can be used as pH sensor.
Research Article
A multi-responsive diarylethene chemosensor via salicylaldehyde linking fluorescein unit
https://doi.org/10.21203/rs.3.rs-2244815/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
multi-responsive
Cu2+
pH
diarylethene
fluorescein
Fluorescence sensors have been gaining increasing attention for recognizing and measuring transition metal ions due to their easy use, good sensitivity, and good responsivity.[1, 2] Particularly, fluorescent sensors for identifying Cu2+ have been highly valued because Cu2+ play a great role in environmental, biological, and chemical systems.[3, 4] For example, copper-dependent enzymes act as catalysts in biochemical reactions providing energy for converting melanin to skin pigmentation. Copper also assists the formation of crosslinks in collagen, thereby maintaining and mending connective tissue.[5–7] However, excessive copper is toxic and will induce neurodegenerative diseases, including familial amyotrophic lateral sclerosis, Menkes and Wilson's disease, prion diseases and Alzheimer's disease.[8] Therefore, it is important to design a fluorescent sensor that specifically recognizes Cu2+.
Photochromism is defined as the reversible transformation of a chemical substance between two forms with distinct absorption spectra by absorbing electromagnetic radiation energy.[9, 10] Specially, diarylethene derivatives are the most promising optical derivatives due to their significant thermally irreversible photochromic behavior and remarkable fatigue resistance.[11, 12] In the photochromic process, a reversible change of color between a colorless state and a colored state upon irradiating different light.[13] For diarylethene-based sensors, the colored state is convenient be to applied in naked-eyes recognition for different analytes, such as metal ions,[14–16] free radicals,[17] and amino acids.[18] Now, some diarylethene-based sensors for detecting different analytes have been reported, but there are a few examples can be used as naked-eyes sensors.
As a kind of fluorophore, fluorescein and its derivatives have attracted great attention due to their prominent photophysical properties including high absorption, excellent fluorescent quantum yield, and remarkable photostability.[19, 20] Fluorescein is not fluorescent at its original state, but will have extraordinary fluorescence (open-ring) and distinct color change upon adding metal ions.[21] Fluorescein-based probes have been previously reported to identify metal ions including Cu2+,[22] Hg2+,[23] and Zn2+.[24] Whereas, fluorescein-based diarylethene sensors have been scarcely studied. In this work, a new diarylethene (1O) containing fluorescein was synthesized. After photochromism, the color of the closed-ring diarylethene (1C) can change from purple to colorless upon adding appropriate amount of Cu2+. Meanwhile, 1C can also be used to monitor the pH of the environment by color change. The absorption and fluorescent properties of 1C were studied systematically. This work provides a novel direction to develop fluorescein-based diarylethene sensors.
2.1. General methods
Chemical reagents were used without further purification after purchase. All solvents were spectroscopic grade and purified with distillation prior to use. Solutions containing metal ions were obtained by dissolving their nitrates (0.1 mmol) in distilled water (10 mL) except K+, Mn2+, Ba2+ and Hg2+ (chlorides were used for these metals). UV/vis spectra and fluorescence spectra were recorded on an Agilent 8453 UV/vis spectrophotometer and a Hitachi F-4600 fluorescence spectrophotometer, respectively. NMR spectra were measured on a Bruker AV400 (400 MHz) spectrometer with CDCl3 and DMSO-d6 as solvents and tetramethylsilane (TMS) as an internal standard. Mass spectra were obtained on an Agilent 1100 ion trap MSD spectrometer. Elemental analysis was carried out with a PE CHN 2400 analyzer.
2.2. Methods
A stock solution of 1O (1.0 × 10− 3 mol L− 1) was prepared in CH3CN. During the testing process of detecting metal ions ion, the 2 mL of 1O solution (2.0 × 10− 5 mol L− 1) in CH3CN was irradiated UV light to form 1C firstly, the fluorescence and absorption were measured by adding appropriate amount of metal ions at room temperature.
2.3. Synthesis of 1O
Figure 1 shows the procedure for synthesizing compound 1O. Compound 2[25–27] and compound 6[28] were obtained according to previous reports.
Compound 3 (1.22 g, 10.00 mmol) and 3-bromoprop-1-yne (1.04 g, 10.00 mmol) were dissolved in 20 mL acetonitrile, potassium carbonate (2.76 g, 20.00 mmol) and potassium iodide (1.65 g, 10.00 mmol) were added and then heated to reflux for 24 hours. After the solution was cooled to room temperature, the precipitate was filtered and 50 mL dichloromethane was added, the solution was washed three times with saturated sodium chloride solution. the dichloromethane was removed under reduced pressure and the product was purified with column chromatography using ether/ethyl acetate (v/v = 4:1) as an eluent to get compound 4 (1.26 g, 8.63 mmol) with 86% yield. 1H NMR (400 MHz, DMSO-d6), δ (ppm): 3.67 (s, 1H, −C ≡ H), 5.04 (s, 2H, −CH2−), 7.14 (t, 1H, −PhH), 7.30 (d, 1H, J = 8.0 Hz, −PhH), 7.70 (q, 2H, −PhH), 10.36 (s, 1H, −CHO) (Fig. S1); ESI-MS (m/z): 161.12 [3 + H+]+; Anal. calcd for C10H8O2: C, 74.99; H, 5.03; O, 19.98%. Found: C, 74.82; H, 5.11; O, 20.07%.
To a stirring 20 mL THF solution of compound 2 (0.40 g, 0.80 mmol) and compound 4 (0.13 g, 0.80 mmol), 2 mL aqueous solutions containing anhydrous copper sulfate (0.31 g, 1.20 mmol) and sodium ascorbate (0.48 g, 2.40 mmol) were added. Then, the solution was sequentially stirred for 24 hours. Subsequently, 1 mL of ammonium hydroxide was added into the solution and kept for stirring for another half an hour. After the completion of reaction, the solvent was removed under reduced pressure and the product was purified with column chromatography using ether/ethyl acetate (v/v = 6:1) as an eluent to obtain the compound 5 (0.38 g, 0.58 mmol) with 73% yield. 1H NMR (400 MHz, DMSO-d6), δ (ppm): 1.83 (s, 3H, −CH3), 1.89 (s, 3H, −CH3), 5.33 (s, 2H, −CH2−), 5.82 (s, 2H, −CH2−), 7.10 (s, 1H, thiophene − H), 7.23 (s, 1H, thiophene − H), 7.31 (t, 1H, −PhH), 7.40 (t, 4H, −PhH), 7.59 (d, 2H, J = 8.0 Hz, −PhH), 7.67 (q, 2H, −PhH), 8.34 (s, 1H, −CH=), 10.31 (s, 1H, −CHO) (Fig. S2); ESI-MS (m/z): 660.62 [5 + H+]+; Anal. calcd for C32H23F6N3O2S2: C, 58.26; H, 3.51; N, 6.37; O, 4.85%. Found: C, 58.17; H, 3.58; N, 6.22; O, 4.97%.
Compound 5 (0.20 g, 0.30 mmol) and compound 6 (0.10 g, 0.30 mmol) were dissolved in 10 mL ethyl alcohol and then heated to reflux for 24 hours. A pale pink precipitate was formed when the solution was cooled to room temperature. The crude product was washed three times with hot ethyl alcohol and filtered to give 1O (0.15 g, 0.15 mmol, yield: 50%). 1H NMR (500 MHz, DMSO-d6), δ (ppm): 1.82 (s, 3H, −CH3), 1.89 (s, 3H, −CH3), 5.10 (s, 2H, −CH2−), 5.89 (s, 2H, −CH2−), 6.38–6.43 (m, 4H, −PhH), 6.54 (s, 2H, thiophene − H), 6.89–6.93 (t, 1H, J = 8.0 Hz, −PhH), 7.05–7.07 (d, 1H, J = 8.0 Hz, −PhH), 7.14–7.16 (d, 1H, J = 8.0 Hz, −PhH), 7.29–7.32 (m, 3H, −PhH), 7.37–7.46 (m, 4H, −PhH), 7.56–7.60 (m, 4H, −PhH), 7.85–7.87 (d, 1H, J = 8.0 Hz, −PhH), 8.17 (s, 1H, −CH=), 9.13 (s, 1H, −CH=), 9.87 (s, 2H, −OH) (Fig. S3). 13C NMR (126 MHz, DMSO-d6), δ (ppm): 164.02, 158.95, 158.80, 156.96, 152.59, 151.05, 143.97, 143.62, 143.20, 142.20, 141.63, 137.17, 134.41, 132.91, 132.15, 129.71, 129.50, 129.45, 128.63, 128.39, 127.66, 125.73, 125.40, 125.19, 124.75, 124.23, 123.99, 123.54, 123.25, 122.86, 121.52, 113.64, 112.66, 110.34, 102.92, 65.56, 61.95, 47.78, 14.48, 14.34 (Fig. S4); ESI-MS (m/z): 849.42 [1O + Na+]+; Anal. calcd for C48H55N7O6: C, 69.80; H, 6.71; N, 11.87; O, 11.62%. Found: C, 69.67; H, 6.88; N, 11.78; O, 11.67%.
3.1 Photochromic properties of diarylethene 1O
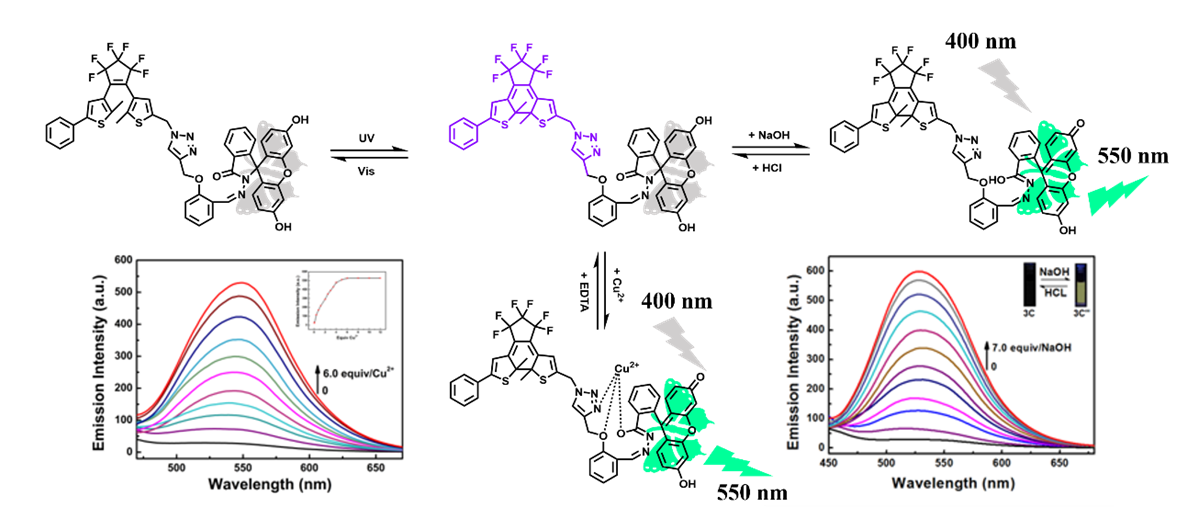
The photochromic properties of diarylethene 1O were evaluated in acetonitrile solution (2.0 × 10− 5 mol L− 1) at room temperature. As shown in Fig. 3A, in the initial state, the open-ring of 1O solution was colorless, and the maximum absorption peak was observed at 279 nm due to a π*-π transition.[29] When being irradiated with a UV light at 297 nm, the color of 1O varied from colorless to purple with a new absorption peak appeared at 555 nm, indicating the formation of a closed-ring 1C. When being irradiated with a visible light (λ > 500 nm), the color of solution disappeared and the absorption spectra regained its original state. The photochromic process between 1O and 1C is shown in Fig. 2. The 1H NMR and 13C NMR of 1C were shown in Figure S5 and S6.Using 1, 2-bis (2-methyl-5-phenyl-3-thienyl) perfluorocyclopentene as a reference, the cyclic and cyclorrhaphous quantum yields were measured to be 0.32 and 0.09, respectively. The fatigue resistance of 1O was tested by alternating UV and visible light irradiation, only 12% degradation of 1O after ten decolorization cycles (Fig. 3B)
3.2 Spectral response of 1C to Cu2+
The selectivity of 1C (2.0 × 10− 5 mol L− 1) to multifarious metal ions (Cu2+, Hg2+, Ca2+, Pb2+, Mn2+, Ni2+, Co2+, Cd2+, Zn2+, Ba2+, Mg2+, Fe3+, Cr3+, Al3+, K+, and Na+) was first tested in acetonitrile solutions at room temperature. As demonstrated in Fig. 4A, only the addition of Cu2+ (6.0 equiv) remarkably decreased the absorbance in visible region of 1C, while other metal ions did not induce much absorption spectral change. Similarly, fluorescence spectral changes of 1C with multifold metal ions (6.0 equiv) were also obtained under the same conditions. The fluorescent emission intensity at 550 nm of 1C solution was only prominently enhanced by the addition of Cu2+ (Fig. 4B). To further attest the high selectivity of 1C over Cu2+, competitive tests were next performed by adding a variety of metal ions to the 1C solution. As shown in Figs. 4C and 4D, there was no interference of other metal ions with 1C for detecting Cu2+. These data prove that compound 1C having highly selective for detecting Cu2+.
In order to further understand the chemosensing characteristics of 1C (2.0 × 10− 5 mol L− 1) to Cu2+, the absorption and fluorescence spectral titration experiments were carried out in acetonitrile solutions at room temperature. As shown in Fig. 5A, the absorbance at 555 nm gradually declined when adding Cu2+ from 0 to 6.0 equiv. At the same time, the color of 1C solution gradually changed from purple to colorless. In addition, a new fluorescent emission band centered at 550 nm gradually intensified when adding Cu2+ from 0 to 6.0 equiv (Fig. 5B). And it is interesting to find that the absorption and fluorescence peaks of 1C were able to resume their original states by adding EDTA (10.0 equiv), indicating the reversibility of 1C binding with Cu2+.
To further understand the binding of 1C and Cu2+, the Job's plot was used to determine the binding ratio using fluorescent titration data.[30, 31] As exhibited in Fig. 6A, the fluorescence intensity reached the maximum when the molar fraction of [Cu2+]/([Cu2+] + [1C]) was equal to 0.5, corresponding to the binding ratio between Cu2+ and 1C is 1:1 in acetonitrile solution. The binding constant of Cu2+ with 1C was determined to be (1.3188 ± 0.41) × 105 mol L− 1 by using the Hildebrand-Benesi equation. (Fig. 6B), and the detection limit was calculated to be 0.11 µM (Fig. 6C).[32] Compared with the reported sensors analysis of Cu2+ (Table 1), the sensor not only showed excellent detection limit, but also exhibited multi-function for the detection of Cu2+.
Table 1 Comparative study of the analytical performance of 1O with other reported sensors.

3.3 Spectral response of 1C to base
As we all know, fluorescein is very sensitive to base due to the presence of two hydroxyl groups in the molecular, which resulted in fluorescein display unique absorption and fluorescence spectra upon the pH changes of its solution. As expected, when 7.0 equiv of NaOH was gradually added into the solution of 1C in acetonitrile at room temperature, the absorption spectra exhibited notable change (Fig. 7A), a new absorption band in the range of 303–450 nm emerged and visible absorption band in the range of 450–675 nm disappeared, the color of solution changed from purple to pale yellow. When the amount of NaOH was more than 4.0 equiv, the absorbance at 555 nm reached a plateau upon further titration (Fig. 7B). Meanwhile, the fluorescent spectra also displayed remarkable change after NaOH was added. As shown in Fig. 7C, the fluorescence at 528 nm notably increased with an evident color change from dark to bright yellow, which ascribed to the formation of open-ring quinoid form of fluorescein moiety. When 5.0 equiv of NaOH was added into the non-fluorescence solution of 1C, the fluorescence intensity at 528 nm reached maximum level (Fig. 7D). The absolute fluorescent quantum yield of the deprotonated 1C was determined to be 0.24. The gradual back addition of excess HCl regenerated the lactone form of fluorescein, while the absorption and fluorescence spectra dropped back to the initial state, suggesting that 1C bearing can as a potential naked-eyes sensor to measure the pH of the environment.
A new diarylethene derivative (1O) has been synthesized by connecting a fluorescein via a salicylaldehyde linkage. The photochromic properties of the compound were studied in acetonitrile solution at room temperature. Compound 1C has a high selectivity and sensitivity for detecting Cu2+. When adding Cu2+ into 1C solution, the color of the solution turned from purple to colorless and the fluorescence changed from dark to yellow. Meanwhile, Compound 1C provides a new clue for monitoring the pH of the environment by naked-eyes method.
Ethical Approval: Not applicable
Competing interests: No conflict of interest to declare
Authors' contributions:
Xue Li. Writing-original draft, Editing
Yin Ai. Investigation
Huimin Kang. Writing-original draft
Congbin Fan. Investigation
Gang Liu. Supervision, Review
Haichang Ding. Editing, Review
Shouzhi Pu. Resources, Supervision
Funding: The authors are grateful for the financial support from the National Natural Science Foundation of China (41867052, 41867053, 22263005) and the Project of the Science Funds of Jiangxi Education Office (GJJ190613, GJJ211135).
Availability of data and materials: Not applicable
- Chen X, Wang F, Hyun JY, Wei T, Qiang J, Ren X, Shin I, Yoon J, (2016) Recent progress in the development of fluorescent, luminescent and colorimetric probes for detection of reactive oxygen and nitrogen species. Chemical Society Reviews 45, 2976-3016. https://pubs.rsc.org/en/content/articlelanding/2016/cs/c6cs00192k
- Stennett EMS, Ciuba MA, Levitus M, (2014) Photophysical processes in single molecule organic fluorescent probes. Chemical Society Reviews 43, 1057-1075. https://pubs.rsc.org/en/content/articlelanding/2014/cs/c3cs60211g
- Chakraborty D, Singh R, (2017) Novel fluorescent vanadylmoxifloxacinato complexes as sensors for Cu2+. Journal of Photochemistry and Photobiology A: Chemistry 335, 211-216. https://www.sciencedirect.com/science/article/abs/pii/S1010603016307067
- Duan G, Zhang G, Yuan S, Ji R, Zhang L, Ge Y, (2019) A pyrazolo[1,5-a]pyridine-based ratiometric fluorescent probe for sensing Cu2+ in cell. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 219, 173-178. https://www.sciencedirect.com/science/article/abs/pii/S138614251930438X
- Z. Zhang, H. Liu, S. Pu, (2018) A highly selective fluorescent chemosensor for Cu2+ based on a diarylethene with a 2,1,3‐benzoxadiazole unit. Journal of Physical Organic Chemistry 31, e3839. https://onlinelibrary.wiley.com/doi/full/10.1002/poc.3839
- Yang JF, Zou Q, Chen XY, Li YZ, Zhao CR, Weng TY, Wu B, Zhu LL, Wang DL, Xin ZL, (2021) The stepwise photochromic reactivity of diarylethene tuned by selective ions and fabrication of a molecular logic circuit. Dyes and Pigments 191, 109361. https://www.sciencedirect.com/science/article/abs/pii/S014372082100228X
- Huang Y, Li CF, Shi WJ, Tan HY, He ZZ, Zheng L, Liu F, Yan JW, (2019) A near-infrared BODIPY-based fluorescent probe for ratiometric and discriminative detection of Hg2+ and Cu2+ ions in living cells. Talanta 198, 390-397. https://www.sciencedirect.com/science/article/abs/pii/S0039914019301444
- Yuan L, Lin W, Chen B, Xie Y, (2012) Development of FRET-Based Ratiometric Fluorescent Cu2+ Chemodosimeters and the Applications for Living Cell Imaging. Organic Letters 14, 432-435. https://pubs.acs.org/doi/full/10.1021/ol202706k
- Chen XY, Zhu LL, Wang DL, Zou Q, Li X, Chen WB, (2019) A unimolecular platform based on diarylethene with multiple stimuli-gated photochromism. Dyes and Pigments 164, 91-96. https://www.sciencedirect.com/science/article/abs/pii/S0143720818326421
- Ding H, Li B, Pu S, Liu G, Jia D, Zhou Y, (2017) A fluorescent sensor based on a diarylethene-rhodamine derivative for sequentially detecting Cu2+ and arginine and its application in keypad lock. Sensors and Actuators B: Chemical 247, 26-35. https://www.sciencedirect.com/science/article/abs/pii/S0925400517303994
- Irie M, Fukaminato T, Matsuda K, Kobatake S, (2014) Photochromism of Diarylethene Molecules and Crystals: Memories, Switches, and Actuators. Chemical Reviews 114, 12174-12277. https://pubs.acs.org/doi/full/10.1021/cr500249p
- van de Linde S, Sauer M, (2014) How to switch a fluorophore: from undesired blinking to controlled photoswitching. Chemical Society Reviews 43, 1076-1087. https://pubsrsc.53yu.com/en/content/articlelanding/2013/cs/c3cs60195a/unauth
- Wang S, Ding H, Wang Y, Fan C, Liu G, Pu S, (2019) Novel multi-responsive fluorescence switch for Hg2+ and UV/vis lights based on diarylethene-rhodamine derivative. Tetrahedron 75, 1517-1524. https://www.sciencedirect.com/science/article/abs/pii/S0040402019301255
- Mao L, Li X, Ding H, Fan C, Liu G, Pu S. (2022) A highly selective Hg2+ fluorescent chemosensor based on photochromic diarylethene with quinoline unit. Journal of Fluorescence, DOI: https://doi.org/10.21203/rs.3.rs-1282041/v1.
- Hu XG, Li XL, Kim SH, Ahn KH, Yang SI, (2020) Gated photochromic reactivity of azadithiacrown-ether functionalized diarylethene. Dyes and Pigments 172, 107869. https://www.sciencedirect.com/science/article/abs/pii/S0143720819317061
- Li X, Li X, Zhao H, Kang H, Fan C, Liu G, Pu S, (2021) A novel diarylethene‑rhodamine unit based chemosensor for fluorimetric and colorimetric detection of Hg2+. Journal of Fluorescence 31, 1513-1523. https://linkspringer.53yu.com/article/10.1007/s10895-021-02775-4
- Xiong KT, Li ML, Jiang Y, Xu HB, Zeng MH, (2020) Imidazole diarylethene switches: an alternative to acid-gated photochromism. New Journal of Chemistry 44, 8061-8067. https://pubsrsc.53yu.com/en/content/articlelanding/2020/nj/d0nj00606h/unauth
- Zhu ZF, Wang YS, Ding HC, Fan CB, Tu YY, Liu G, Pu SZ, (2021) A novel full symmetric diarylethene-based ratiometric fluorescent sensor for lysine and the application for a logic circuit. Luminescence 36, 691-697. https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/full/10.1002/bio.3988
- Lee CH, Yoon DW, Sessler JL, (2008) Strapped and other topographically nonplanar calixpyrrole analogues. Improved anion receptors. Chemical communications 24-34. https://pubsrsc.53yu.com/en/content/articlehtml/2008/cc/b713183f
- Yoon J, Kim SK, Singh NJ, Kim KS, (2006) Imidazolium receptors for the recognition of anions. Chemical Society Reviews 35, 355-360. https://pubsrsc.53yu.com/en/content/articlelanding/2006/cs/b513733k/unauth
- Wang D, Ren AM, Guo JF, Zou LY, Huang S, (2015) Computational design of a two-photon excited FRET-based ratiometric fluorescent Cu2+ probe for living cell imaging. RSC Advances 5, 98144-98153. https://pubsrsc.53yu.com/en/content/articlelanding/2015/ra/c5ra18393f/unauth
- Chen B, Wang LL, Zhao YF, Ni Y, Xin CQ, Zhang CW, Liu JH, Ge JY, Lin L, Huang W, (2017) Photocontrollable fluorogenic probes for visualising near-membrane copper (ii) in live cells. RSC Advances 7, 31093-31099. https://pubsrsc.53yu.com/en/content/articlehtml/2017/ra/c7ra03559d
- Li L, Wang CY, Wu JJ, Tse YC, Cai YP, Wong KMC, (2015) A molecular chameleon with fluorescein and rhodamine spectroscopic behaviors. Inorganic Chemistry 55, 205-213. https://pubs.acs.org/doi/full/10.1021/acs.inorgchem.5b02147
- Li DL, Liu L, Li WH, (2015) Genetic targeting of a small fluorescent zinc indicator to cell surface for monitoring zinc secretion. ACS Chemical Biology 10, 1054-1063. https://pubs.acs.org/doi/full/10.1021/cb5007536
- Ding HC, Liu G, Zheng CH, Pu SZ, (2014) Multi-addressable fluorescent switch based on a photochromic diarylethene with triazole-bridged methylquinoline group. Dyes and Pigments 103, 82-88. https://www.sciencedirect.com/science/article/abs/pii/S014372081300466X
- Xu HT, Ding HC, Li G, Fan CB, Liu G, Pu SZ, (2017) A highly selective fluorescent chemosensor for Fe3+ based on a new diarylethene with a rhodamine 6G unit. RSC Advances 7, 29827-29834. https://pubsrsc.53yu.com/en/content/articlehtml/2017/ra/c7ra04728b
- Xia SJ, Liu G, Pu SZ, (2015) A highly selective fluorescence sensor for Zn2+ and Cu2+ based on diarylethene with a piperazine-linked amidoquinoline unit. Journal of Materials Chemistry C 3, 4023-4029. https://pubsrsc.53yu.com/en/content/articlelanding/2015/tc/c5tc00315f/unauth
- Duan F, Liu G, Fan C, Pu SZ, (2016) Synthesis and photochromism of a novel amphiphilic diarylethene bearing two cholic acid groups. Tetrahedron Letters 57, 1963-1966. https://www.sciencedirect.com/science/article/abs/pii/S004040391630301X
- Li ZX, Liao L, Sun W, Xu CH, Zhang C, Fang CJ, Yan CH, (2008) Reconfigurable cascade circuit in a photo-and chemical-switchable fluorescent diarylethene derivative. Journal of Physical Chemistry C 112, 5190-5196. https://pubs.acs.org/doi/full/10.1021/jp711613y
- Yang XB, Ge JF, Xu YJ, Xu QF, Liang J, Lu JM, (2011) Benzo[a]phenoxazinium-Based Red-Emitting Chemosensor for Zinc Ions in Biological Media. Organic Letters 13, 2710-2713. https://pubs.acs.org/doi/full/10.1021/ol2008022
- Kato R, Nishizawa S, Hayashita T, Teramae N, (2001) A thiourea-based chromoionophore for selective binding and sensing of acetate. Tetrahedron Letters 42, 5053-5056. https://www.sciencedirect.com/science/article/abs/pii/S0040403901009169
- Benesi HA, Hildebrand JHJ, (1949) A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons. Journal of the American Chemical Society 71, 2703-2707. https://pubs.acs.org/doi/pdf/10.1021/ja01176a030
- Huang Q, Chen YT, Ren YW, Wang ZY, Zhu YX, Zhang Y, (2018) A rapid and naked-eye visible rhodamine 6G-based chemosensor for sensitive detection of copper(ii) ions in aqueous solution. Analytical Methods 10, 5731-5737. https://pubsrsc.53yu.com/en/content/articlelanding/2018/ay/c8ay02019a/unauth
- Lu X, Wang W, Dong Q, Bao X, Lin X, Zhang W, Dong X, Zhao W, (2015) A multi-functional probe to discriminate Lys, Arg, His, Cys, Hcy and GSH from common amino acids. Chemical Communications 51, 1498-1501. https://pubsrsc.53yu.com/en/content/articlelanding/2014/cc/c4cc07757a/unauth
- Feng E, Lu R, Fan C, Zheng C, Pu S. (2017) A fluorescent sensor for Al3+ based on a photochromic diarylethene with a hydrazinobenzothiazole Schiff base unit. Tetrahedron Letters 58, 1390-1394. https://www.sciencedirect.com/science/article/abs/pii/S0040403917302460
- You GR, Park GJ, Lee JJ, Kim C. (2015) A colorimetric sensor for the sequential detection of Cu2+ and CN− in fully aqueous media: practical performance of Cu2+. Dalton Transactions 44, 9120-9129. https://pubsrsc.53yu.com/en/content/articlelanding/2015/dt/c5dt00772k/unauth
- Arabahmadi R. (2019) A selective chemosensor and fluorescence probe for relay recognition of cations and fluoride ions in aqueous media with logic gate function. Talanta 194, 119-126. https://www.sciencedirect.com/science/article/abs/pii/S0039914018310440
- Thangaraj A, Bhardwaj V, Sahoo SK. (2019) A multi-analyte selective dansyl derivative for the fluorescence detection of Cu(ii) and cysteine. Photochemical & Photobiological Sciences 18, 1533-9. https://linkspringer.53yu.com/article/10.1039/c9pp00080a
- Yin G, Yao J, Hong S, Zhang Y, Xiao Z, Yu T, (2019) A dual-responsive colorimetric probe for the detection of Cu2+ and Ni2+ species in real water samples and human serum. Analyst 144, 6962-6967. https://pubsrsc.53yu.com/en/content/articlelanding/2019/an/c9an01451a/unauth
- Li L, Li H, Liu G, Pu S. (2017) A colorimetric and fluorescent chemosensor for selective detection of Cu2+ based on a new diarylethene with a benzophenone hydrazone unit. Luminescence 32, 1473-1481. https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/full/10.1002/bio.3347
- Kumar A, Bera A, Kumar S. (2020) A Smartphone-Assisted Sensitive, Selective and Reversible Recognition of Copper Ions in an Aqueous Medium. ChemistrySelect 5, 1020-1028. https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/slct.201904399
No competing interests reported.
- Supportinginformation.docx
- 1.png
A novel multi-responsive diarylethene derivative (1O) by linking a fluorescein group was synthesized and its multi-responsive characteristic was investigated.
{kind=link}