Clinical Spectrum
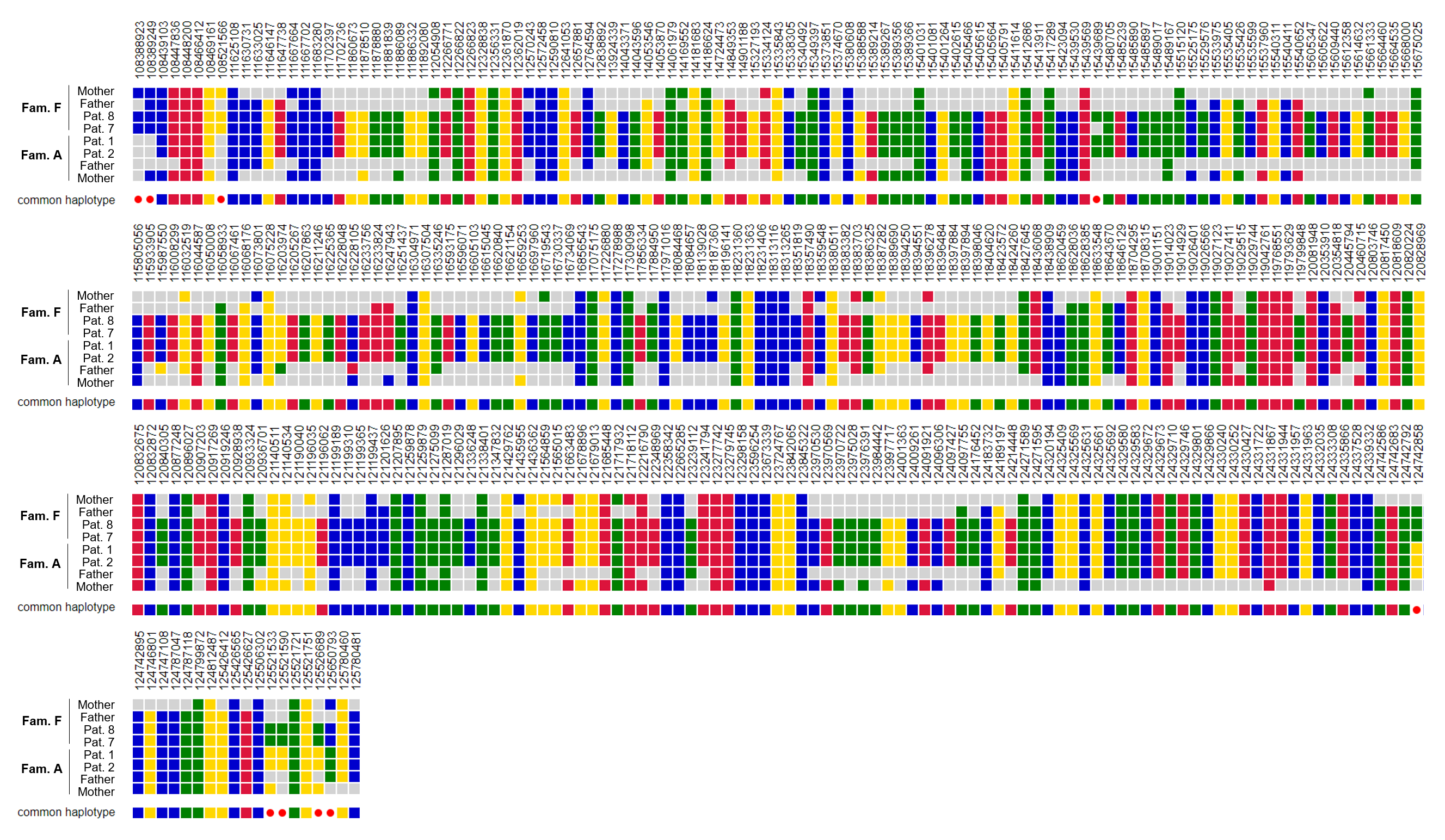
The clinical findings of the ten individuals are summarized in Table 1. Comparison of clinical symptoms of this cohort and previously reported cases are summarized in Fig. 1a. Pedigrees are shown in Fig. 1b and detailed clinical reports are provided in supplementary file 1.
Our cohort of new cases comprised ten individuals (sex distribution 50:50) from eight families. Consanguinity was reported in five of them. All documented birth measurements were within the normal range. Postnatal adaptation and development in the first weeks of life were unremarkable in nine out of ten children. Only one child (individual 5) developed postnatal complications in the form of respiratory distress and pulmonary hypertension. In the following months, recurrent upper airway infections, anemia and hepatomegaly were noted and she died prematurely at two years of age because of viral pneumonia. All other individuals were between 30 months and 19 years of age at last follow-up. Eight out of nine individuals had no respiratory or pulmonary symptoms. At the last clinical examination, all individuals with documented head circumference data were below average and three of them had a head circumference below − 2 SD. Short stature and decreased body weight were observed in four and seven individuals, respectively, with four individuals presenting with both. All ten individuals showed global developmental delay (NDD) and later intellectual disability (ID), which was categorized as moderate or severe in most cases. Behavioral abnormalities in the form of hyperactivity, impulsive, aggressive and autistic behavior were reported in three cases. Eight children were nonverbal at the last examination, and two could speak a few rudimentary words. In three cases, regression of speech occurred after 1–3 years of age. Four individuals never learned to walk unaided, two depended on a wheelchair for longer distances and four had an unstable and broad-based gait. Seizures occurred in seven individuals, with an age of onset between nine months and eight years. In individual 5, epileptiform discharges were documented but no clinical seizure was observed. In four cases, multiple antiepileptic drugs were tried and two children were equipped with a VNS device. Brain abnormalities (dilated ventricles, corpus callosum hypoplasia and mild cortical atrophy) were diagnosed in three individuals, while four had unremarkable brain MRI scans. Other neurological symptoms frequently documented in this cohort included axial hypotonia (9/10) and dystonia (3/10). Furthermore, gastrointestinal problems (3/10) and anemia (3/10) were present in three children, although individual 1 had a confirmed ß-thalassemia.
In summary, all thus far described 23 individuals displayed NDD/ID. In 20 out of 22 axial hypotonia was described and dystonia in 12 of 21. Structural brain abnormalities were present in about 65% of the cases, in which brain MRI was performed. Seizures (or pathological EEG) were documented in 17 of 22 individuals, and 14 of 21 had behavioral abnormalities. Of the children who learned to speak or walk unaided, neuroregression and gait disturbances were described in a subset (4/15 and 6/10, respectively). Approximately half of the reported individuals had recurrent respiratory infections and respiratory distress in the first year of life. Interstitial changes on chest CT were seen in 7 of 10 individuals. Almost half of the children (10/21) had diarrhea. Poor weight gain and/or decreased body weight were described in 15 of 23 cases and one third had short stature or growth retardation.
Molecular Findings
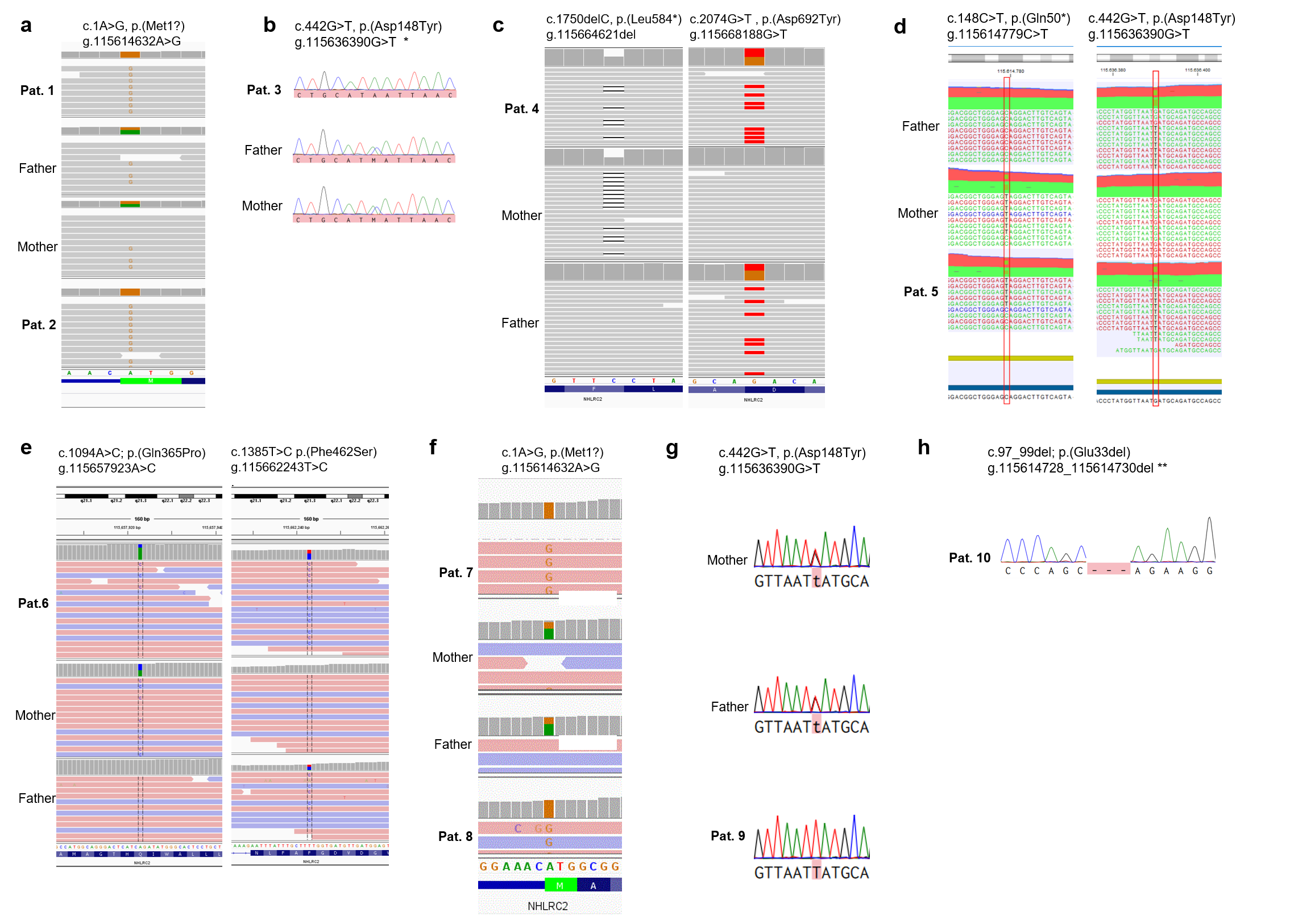
In addition to the recurrent missense variant p.(Asp148Tyr), we identified seven novel variants, including a start-loss variant, three missense variants, a single amino acid in-frame deletion and two nonsense variants. The localization of the variants, the level of conservation and the pathogenicity prediction using different in silico tools are summarized in Fig. 2 and supplementary table 2. Western Blot showed significant reduction of NHLRC2 protein levels in all tested patients samples compared to control samples (Fig. 2d). The results of segregation analysis are illustrated in Supplementary Figure S2.
In an individual with early pulmonary distress (individual 5) the known pathogenic missense variant p.(Asp148Tyr), was detected in trans with an early truncating variant p.(Gln50*). Accordingly, the latter was classified as pathogenic and has already been uploaded to the Clinvar database (ID: 1319311). In two unrelated individuals (3 and 9) without pulmonary symptoms, ES detected the known pathogenic variant p.(Asp148Tyr) in homozygous state.
The NHLRC2 start-loss variant c.1A > G was detected in four individuals (1,2,7 and 8), of two unrelated families. The variant is absent from controls according to the gnomAD database. By analyzing the individual vcf files we confirmed that they share the same disease haplotype (Figure S1). NHLRC2 protein levels, extracted from LCL derived cells of individual 1 and 2, were strongly reduced compared to control samples (Fig. 2d). Since the closest in-frame alternative translation start codon is located at c.433, p.145, we classified this variant according to the ACMG guidelines as likely pathogenic (PVS1_mod, PM2_sup, PM3_sup, PP1_mod, PP4).
In individual 4, a private nonsense variant p.(Leu584*) was detected in combination with the rare missense variant c.2074G > T, p.(Asp692Tyr), which is listed 8x in heterozygous state in gnomAD. The stop-gain variant is predicted to undergo nonsense-mediated decay (NMD) and is thus classified as likely pathogenic (PVS1, PM2_sup). Western Blotting showed significant reduction of NHLRC2 protein levels compared to control samples (Fig. 2d). The missense variant affects a moderately conserved residue located in the ß-strand domain. In silico 3D modeling data of the protein acquired via the MaSIF tool, indicated that the region, which contains the altered residue (Asp692Tyr) might be important for binding interaction partners of NHLRC2 (Fig. 4d). We still classified this variant as a variant of uncertain significance (VUS) (PM2_sup, PM3, PP3, PP4).
Two missense variants [c.1094A > C, p.(Gln365Pro) and c.1385T > C, p.(Phe462Ser)] affecting highly conserved residues within the ß-propeller domain, were detected in compound heterozygous state in individual 6. While the former is listed once in heterozygosity in gnomAD, the latter has an allele frequency of 0.1% in the non-Finnish European population. The insertion of a proline is uniformly predicted as pathogenic by different in silico tools, since proline can disrupt secondary structures like the helix. Interestingly, the second variant appears to affect a predicted binding region of the protein, located in the Thioredoxin-Domain. The near-surface exchange of an amino acid with hydrophobic side chains (Phe) to one with polar side chains (Ser) results in a reduced prediction as a binding site (Fig. 4c). However, applying the ACMG-framework, both variants would still have to be classified as a VUS, until other affected individuals are reported in literature p.(Gln365Pro): PM1_sup, PM2_sup, PP3, PP4; p.(Phe462Ser): PM1_sup, PM3_sup, PP3_mod, PP4.
In individual 10, ES detected a single amino acid in-frame deletion, affecting the highly conserved residue Glu33. The variant is absent from the population database gnomAD. As long as no other affected parties are published with this variant, the variant must remain formally classified as a VUS (PM2_sup, PM3_sup, PM4_sup, PP4).
To assess possible genotype-phenotype correlations we compared variants and their combinations identified in individuals with and without pulmonary affection (Fig. 3a). To further investigate how these NHLRC missense variants impact folding of the protein, we have modeled all NHLRC2 missense variants and the Glu33 deletion with the AlphaFold tool (Fig. 3b-d). For example, in the recurrent missense variant, the replacement of the negatively charged amino acid aspartate by the polar, uncharged threonine presumably results in the loss of several hydrogen bonds (Fig. 3c). Also the majority of the further variants analyzed leads to a loss of at least one hydrogen bond formed at the wildtype position of the respective variant with the only exceptions of the p.(Pro338Leu) and the p.(Gly326Val) variant (Fig. 4a). In order to estimate how the predicted protein structure is altered by the variants in general, we next computed RMSD scores between all missense variants and the deletion variant against the wildtype protein (Fig. 4c). Lower RMSD scores, representing a better fit of backbone atoms between two models, were observed for the p.(Gly326Val) and p.(Pro338Leu) variants. A finding which is in line with hydrogen bond persistence observed in the previous analysis. Consistent with this, Glycine and Valine are both small non-polar amino acids and Proline and Leucine are also non-polar.
Further analyses of the amino acid positions affected by the missense variants with the MaSIFtool suggested possible protein binding sites in proximity to amino acids Phe462 and Asp692. We therefore modeled the respective missense variants and their potential impact on these binding sites. While we could observe a decrease in likelihood of protein-protein interaction at Phe462 for the p.(Phe462Ser) variant, we could not detect such an impact for the p.(Asp692Tyr) variant (Fig. 4c-d).
{kind=link}
{kind=link}