Cell culture
Two immortalized normal human mesothelial cell lines, MeT-5A (pleural mesothelial) and HOMC-D4 (omental mesothelial; intermediate type), and five human mesothelioma cell lines, ACC-MESO-4, Y-MESO-12, Y-MESO-14, Y-MESO-9, and NCI-H2452, were kindly provided by Dr. Y. Sekido, Division of Molecular Oncology, Aichi Cancer Center Research Institute (Nagoya, Japan). The HOMC-D4 cell line was maintained as described previously (35). Y-MESO-9, Y-MESO-12, Y-MESO-14, Y-MESO-9, ACC-MESO-4, and NCI-H2452 cell lines were cultured in RPMI-1640 (Wako, Osaka, Japan) medium containing 10% fetal bovine serum (Sigma) and 1% penicillin-streptomycin (Wako) at 37°C in a 5% CO2 humidified atmosphere.
Gene Knockout Using The Clustered Regularly Interspaced Short Palindromic Repeat (Crispr)/cas9 System
The CRISPR/Cas9 system was used to disrupt the BAP1 gene as described previously (36–37). pSpCas9(BB)-2A-GFP (PX458) was gifted by Feng Zhang (Addgene plasmid # 48138) (36). Briefly, a single guide RNA (sgRNA) sequence was selected using an optimized CRISPR design (http://crispr.mit.edu/). The sgRNA sequence used for BAP1 was 5′- TCAAATGGATCGAAGAGCGC − 3′ and the sequence used for CAMK2D was 5′- CACCGCAGCATATTCTTGTCCAGT − 3′, corresponding to exons 4 and 2, respectively. The plasmid expressing hCas9 and the sgRNA were prepared by ligating the oligonucleotides into the BbsI site of PX458 (BAP1/PX458 and CAMK2D/PX458).
The knockout clones were created by electroporating 1µg of BAP1/PX458 plasmid into 1 × 106 cells using a 4D-Nucleofector™ system (Lonza Japan, Tokyo, Japan). Three days post-transfection, GFP-expressing cells were sorted using BD FACS ARIA III (BD Bioscience). A single clone was selected, expanded, and used for biological assays.
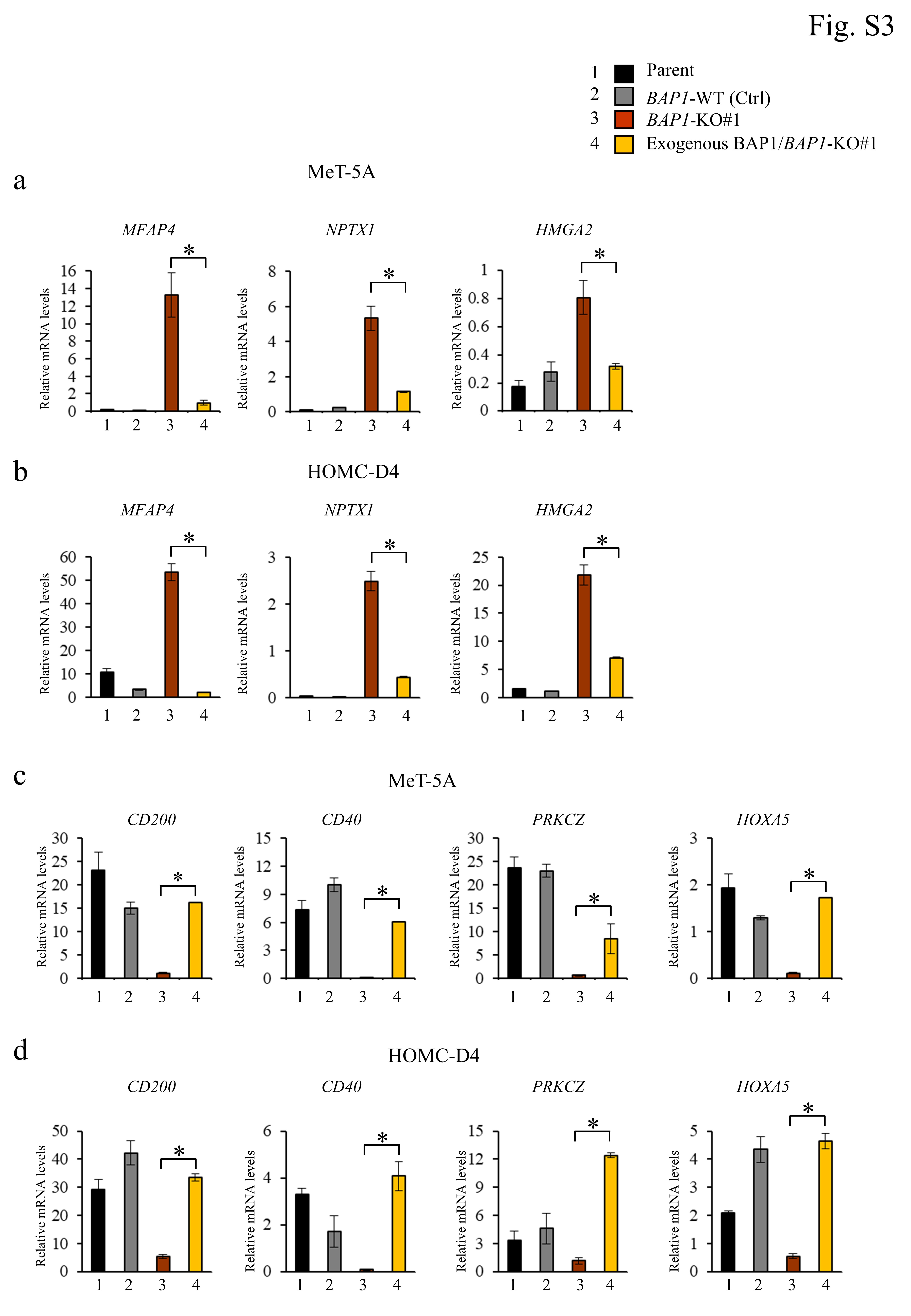
For BAP1 transduction, the BAP1/pcDNA3.1 vector was transfected into a BAP1-KO#1 clone with the 4D-Nucleofector System using the backbone pcDNA3.1 as a control vector. After transfection, the cells were incubated for 48 h, washed with phosphate-buffered saline (PBS), and lysed in the loading buffer. The lysates were used for Western blot analysis to check BAP1 expression.
Quantitative Rt-pcr (Qrt-pcr)
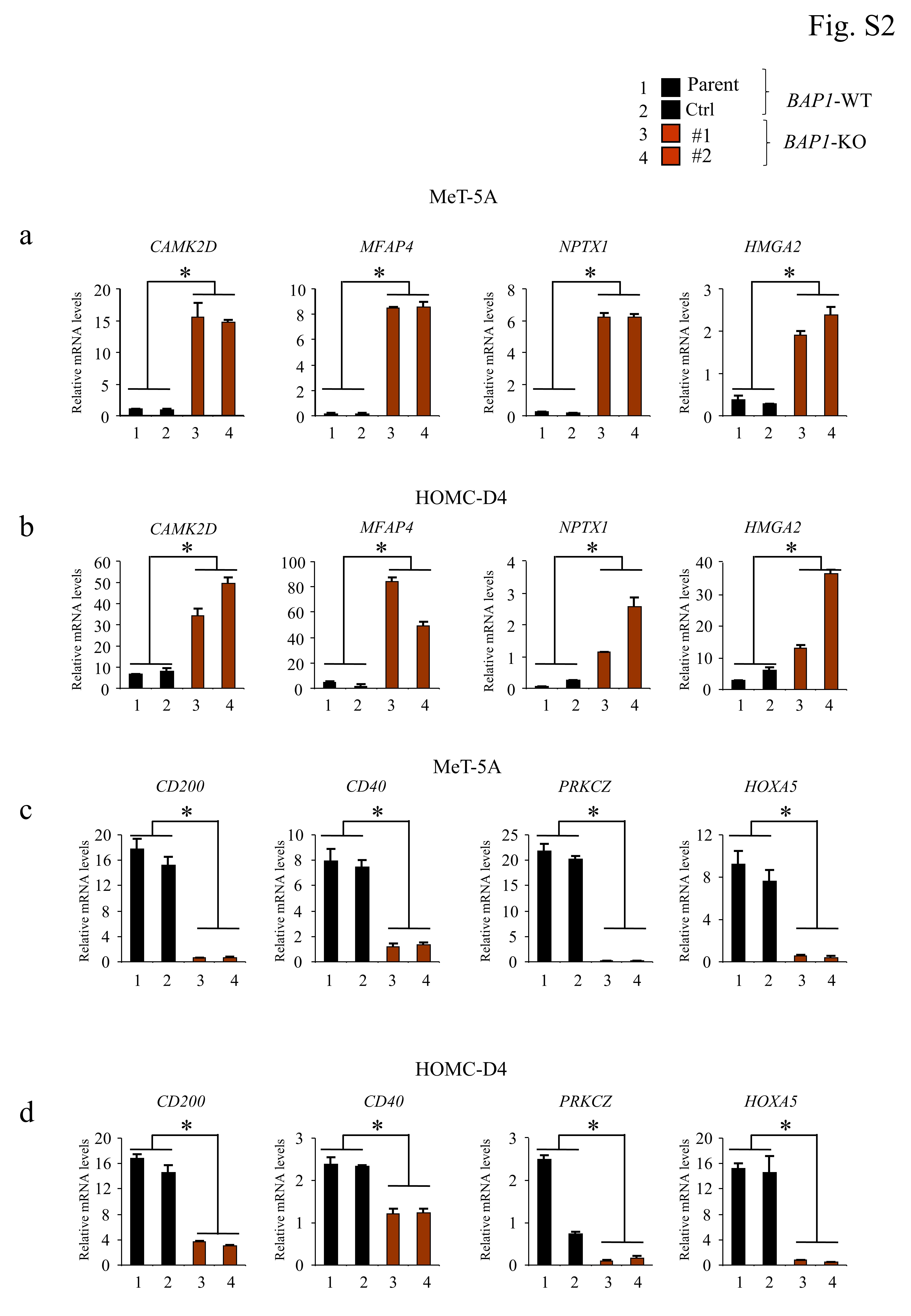
qRT-PCR analysis was performed using SYBR Green I detection as previously described (38) using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal control. The primers used are provided in Table S4.
Cdna Microarray Analysis
cDNA microarray analysis was conducted based on the manufacturer's protocol (Agilent Technologies), as described previously (39). Briefly, we performed cDNA synthesis and cRNA labeling with cyanine 3 (Cy3) dye using the Agilent Low Input Quick Amp Labeling Kit (Agilent Technologies). The Cy3-labeled cRNA was purified, fragmented, and hybridized onto a Human Gene Expression 8x60K v2 Microarray Chip containing 62,969 Entrez Gene RNAs using a Gene Expression Hybridization kit (Agilent Technologies). The raw and normalized microarray data were submitted to the NCBI GEO database (accession number GSE168340; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168340).
Cell Growth Assay
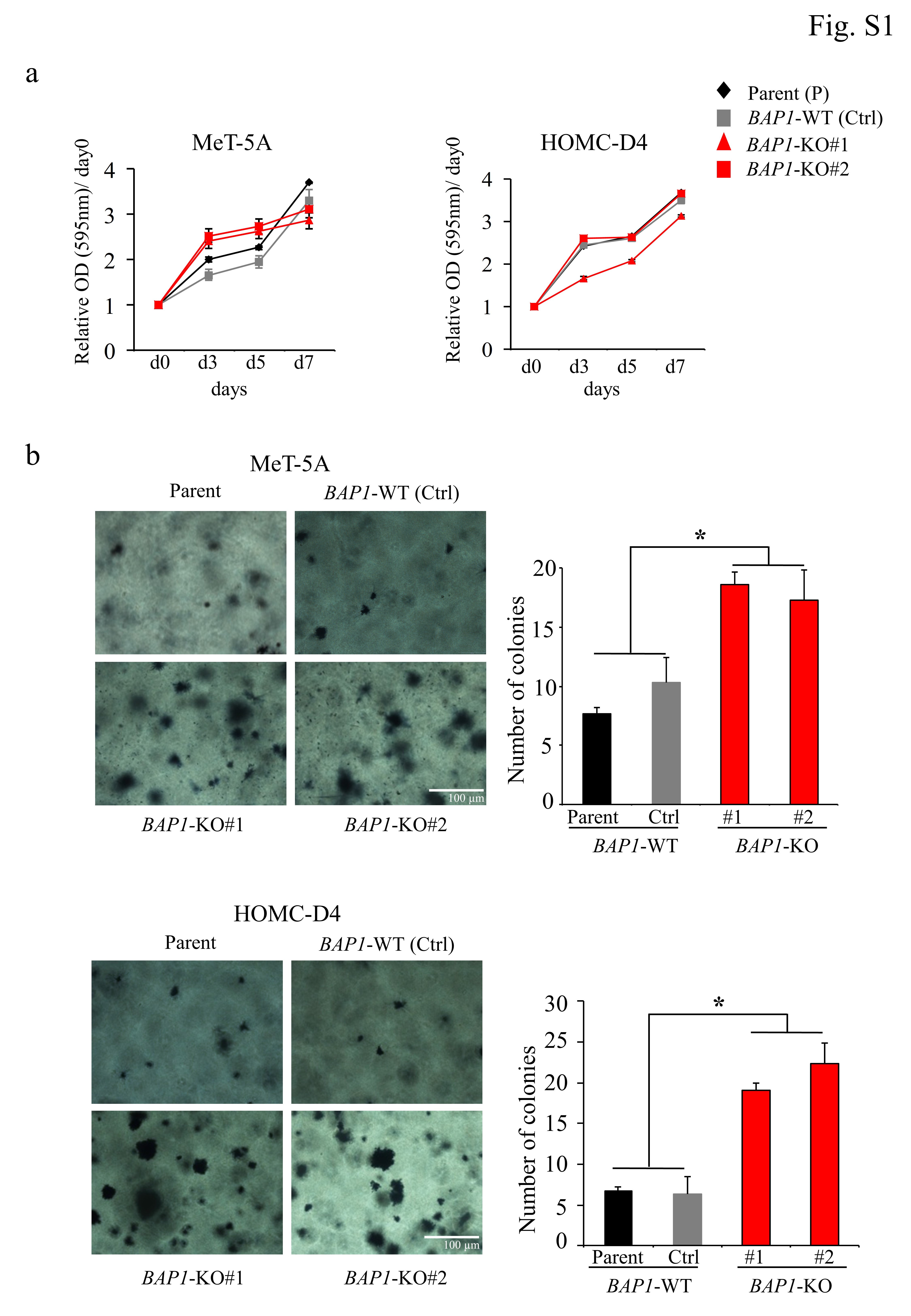
The cell growth rate was measured using MTT assays. Briefly, 1 × 103 cells/well were seeded into 96-well plates and cultured for the indicated times. Subsequently, 10 µl of MTT solution (5 mg/ml; Sigma-Aldrich) was added to each well, and the cells were incubated for 4 h. Next, cell lysis buffer was added to the wells to dissolve the colored formazan crystals. The absorbance was measured at 595 nm using a SpectraMAX M5 spectrophotometer (Molecular Devices, Sunnyvale, CA, USA).
Soft Agar Colony Formation Assay
A soft agar colony formation assay was performed as described previously (15, 16, 40). The number of colonies was counted using Colony Counter software (Keyence, Tokyo, Japan).
Western Blot Analysis
Western blot analysis was performed as described previously (40). The antibodies used are listed in Table S5. Immune complexes were detected using Immuno Star LD (Wako Pure Chemical Industries, Ltd., Osaka, Japan) along with a LAS-4000 image analyzer (GE Healthcare, Tokyo, Japan).
Annexin V Assay
Cells were seeded into 6-well culture plates (5 × 105 cells/ well) and incubated with KN-93 (7.5 µM) for 48 h. After incubation with annexin V (Ax)-FITC and PI (10 µg/mL) at room temperature for 15 min, the fluorescence intensities were measured using fluorescence-activated cell sorting (FACS) using a FACS CantoII (BD, Franklin Lakes, NJ, USA).
Immunohistochemistry (Ihc)
IHC analysis was performed based on a previous method (41) using a human mesothelioma tissue array from US Biomax (MS801b and MS-1001a; Rockville, MD, USA). Sections were incubated with primary antibodies (BAP1 and CAMK2D antibodies, 2 µg/ml). We used either normal rabbit immunoglobulin G or no primary antibody as negative controls. Immunoreactivity was evaluated independently by two investigators (S.K. and H.M.). The staining intensity was scored as strong (+ 3), moderate (+ 2), weak (+ 1), or negative (0).
Screening Of Anticancer Drugs Library
The Screening Committee of Anticancer Drugs (SCADS) library, containing 363 compounds in four 96-well microplates (http://scads.jfcr.or.jp/kit/index.html) was kindly provided by Grant-in-Aid for Scientific Research on Innovative Areas, Scientific Support Programs for Cancer Research, from The Ministry of Education, Culture, Sports, Science and Technology, Japan. The compounds, consisting mostly of anticancer drugs and kinase inhibitors, were originally dissolved at a concentration of 10 mM in dimethyl sulfoxide (DMSO) solution. BAP1-KO and parental MeT-5A cells (3 ×103 cells /well) were seeded into 96-well plates. Next day, cells were treated with a final concentration of 10 µM of each compound or diluent DMSO control (final 0.1% DMSO), and were further incubated for 72 h. MTT assays were performed according to the manufacturer’s instructions. The absorbance was measured at 595 nm using a spectrophotometer. The percentages of cell survival are calculated after normalizing to the mean optical densities in the untreated cells, which were arbitrarily defined as 100%. To identify the compounds with potent antiproliferative effects on BAP1-KO cells, we calculated the differences in the cell survival percentages between BAP1-KO and parental cells.
Xenograft Experiments
The animal experiments were approved by the ethics committee of Aichi Medical University and performed according to the established guidelines. Female Fox Chase SCID mice (CB17/Icr-Prkdcscid/IcrIcoCrl) (6–8 weeks old, each weighing 17–18 g) were purchased from CLEA Japan, Inc (Tokyo, Japan) and bred at the Institute of Animal Experiments in Aichi Medical University in specified pathogen-free animal facilities. Y-MESO-9 cells (1 × 107 cells) were injected subcutaneously into these mice. When the inoculated tumor reached 100 mm3 (day 0), the mice were randomly divided into two groups (treatment and control groups). KN-93 (15 mg/kg body weight) was intraperitoneally administered on days 0, 4, 8, 13, and 16 to each mouse in the treatment group. The control group received PBS as vehicle control. The tumor volume was measured every three to four days and calculated using the modified ellipsoid formula (1/2 × length × width2).
In vivo effects of KN-93 on BALB/c mice
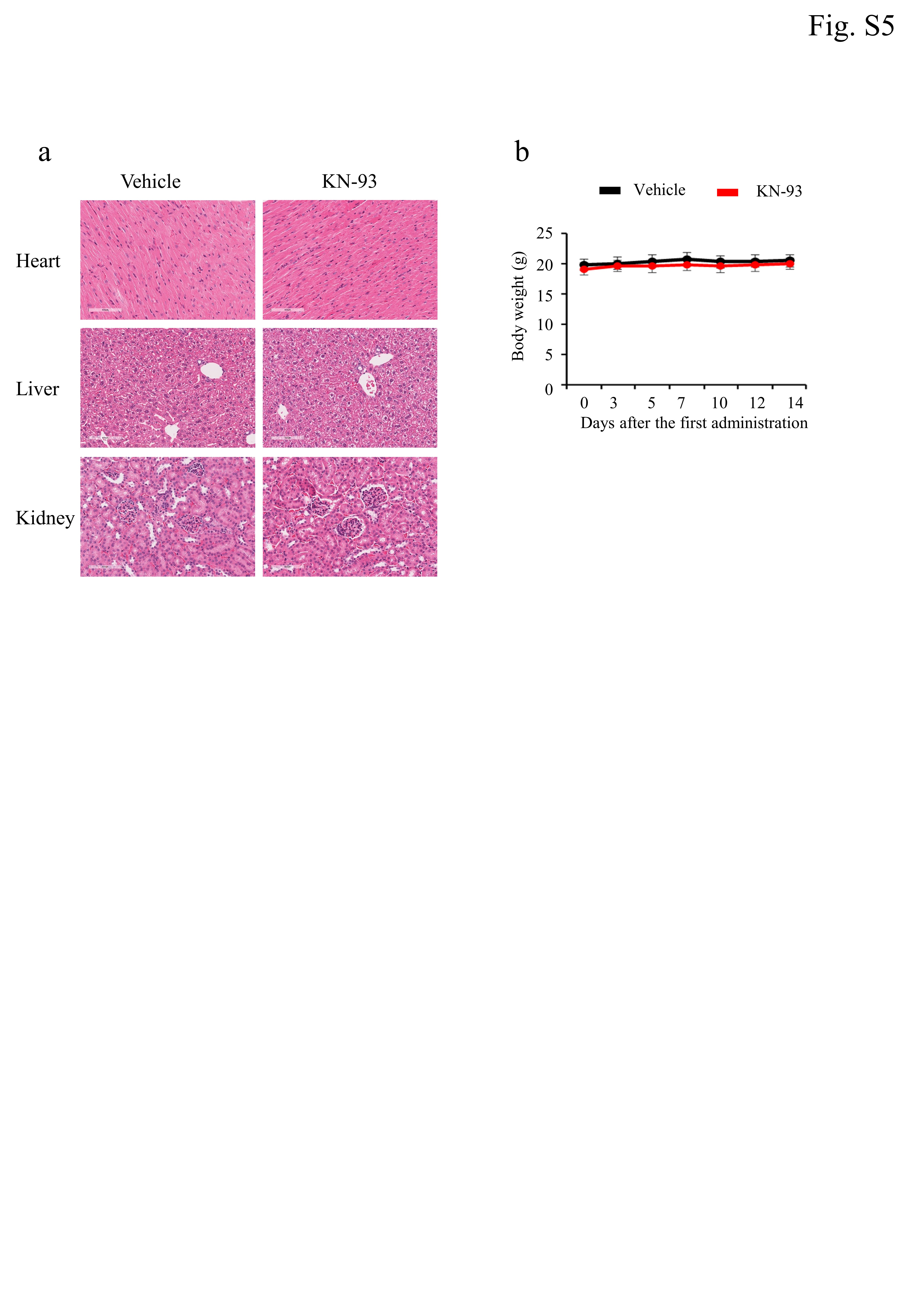
Eight-week-old female BALB/cCrSlc (n = 5, Japan, Inc, Tokyo, Japan) mice were obtained to evaluate the in vivo effects of KN-93. On days 0, 3, 5, 7, 10, and 12, KN-93 (15 mg/kg body weight) or PBS (vehicle control) was intraperitoneally administered to mice. Body weights were measured three times a week. At 14 days after the first administration, the control and treatment group mice were anesthetized using isoflurane, and approximately 0.5–1 ml of blood was collected in a heparin tube. The heparin-plasma samples were examined for blood chemistry by the Nagahama Life Science Laboratory (Oriental Yeast Co., Ltd., Shiga, Japan). The heart, liver, and kidney were carefully dissected for histochemical analyses from mice after the isolation of whole blood. Each organ was washed with PBS and fixed with phosphate-buffered formalin for 24 h. The tissues were trimmed, embedded in paraffin, sectionized into 5-µm thick sections, and stained with hematoxylin and eosin (H&E) (ab245880, Abcam, Cambridge, UK). The stained sections were observed by a microscope (BX41, Olympus, Tokyo, Japan).
Intracellular Ca Assay
Intracellular Ca2+ levels were measured using the CS22 Calcium kit-Fluo 4 (Dojindo, Japan) according to the manufacturer’s protocol. Shortly after removing the medium without damaging the cells, 100 µl of loading buffer was added to each well of 96-well plates. After incubating the cells at 37℃ for 1 h, the loading buffer was replaced with 100 µl warm recording medium (1X) preheated to 37℃. Fluorescence intensity changes were measured at an excitation wavelength of 480–500 nm and emission at 518 nm.
Statistical analysis
The results are expressed as the mean ± SE. Statistical significance between groups was determined using a one-way analysis of variance and Dunnett's comparison. Statistical analyses were performed using SPSS 23.0 software (SPSS Inc, Chicago, Illinois, USA).
{kind=link}
{kind=link}
{kind=link}
{kind=link}