Cell culture
Human gastric epithelial cells (GES-1) and gastric cancer cell lines AGS, SGC7901 were purchased from Cell Bank, Type Culture Collection Committee of Chinese Academy of Sciences (CAMS, Beijing, China). Cells were grown in Dulbecco’s modified essential medium or RPMI1640 supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin at 37 °C in a humidified incubator with 5% CO2. Sophoridine was purchased from MedChemExpress (Shanghai, China) and dissolved in dimethyl sulfoxide (DMSO) to prepare a 10mM stock solution for use.
Cell viability assay
Cell viability in response to Sophoridine treatment was determined using CCK-8 assay (Beyotime, Shanghai, China). In brief, cells seeded in flat bottom 96 well plates (5×103 cells/well) were either treated with Sophoridine at indicated concentrations or treated with indicated drugs for 24 h. Subsequently, medium was discarded, and CCK-8 solution was added, followed by 4 h of incubation. The absorbance was detected by Spectra-Max 190 microplate reader (Molecular Devices) at 450 nm. The percentages of survival cells were measured based on the absorbance of DMSO-treated cells.
EdU assay
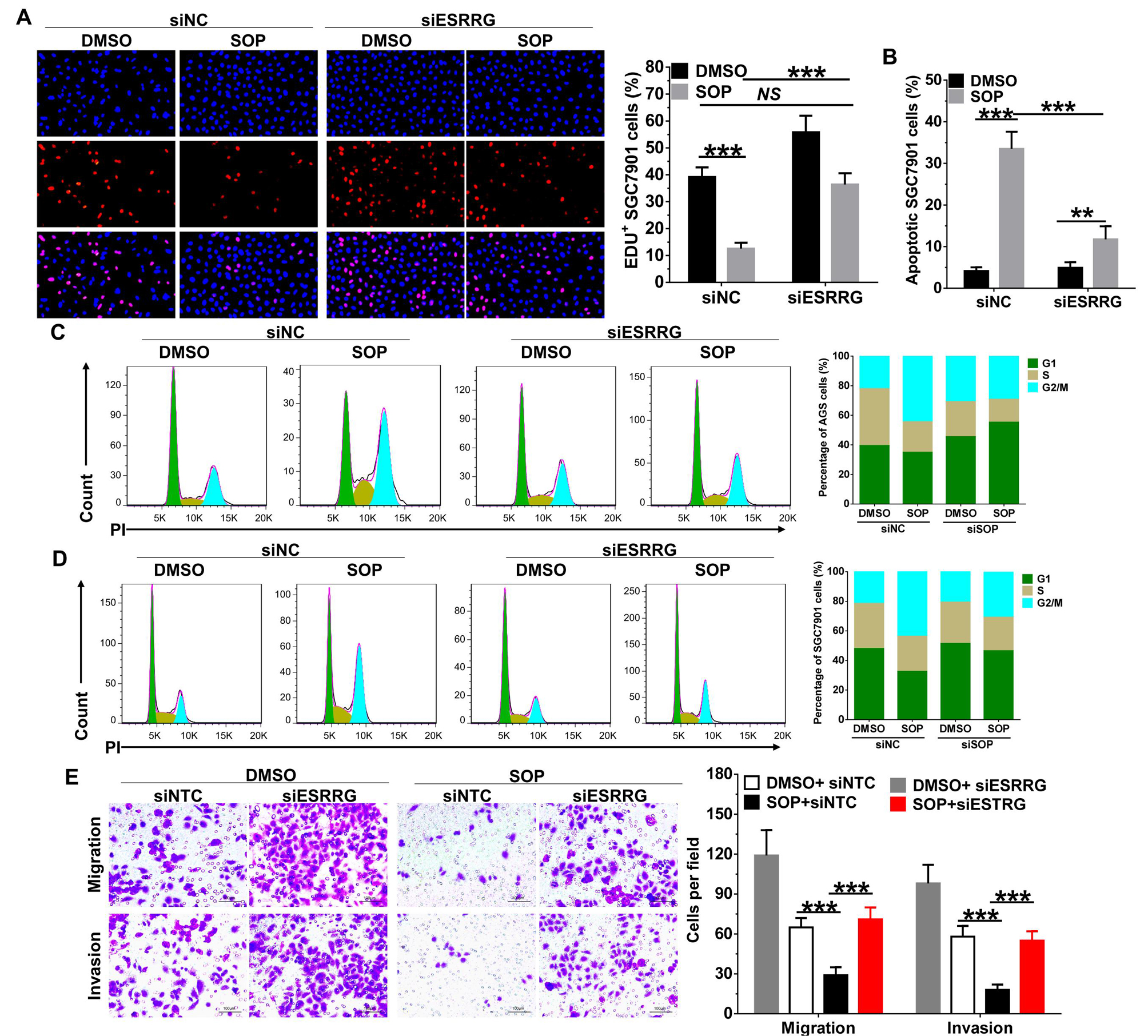
Cell proliferation assay was performed using the BeyoClick™ EdU Cell Proliferation Kit with Alexa Fluor 647 (Beyotime). Briefly, cells with or without transfection were seeded in 96-well plates at a density of 5 × 103 cells/well and then treated with Sophoridine (3 μM). Subsequently, the cells were incubated with 10 μM EdU for 2 h at 37 °C. After being fixed with 4% paraformaldehyde for 30 min, the cells were treated with 0.5% Triton X-100 for 10 min and rinsed with PBS three times. Thereafter, the cells were exposed to 100 μL of click reaction cocktail for 30 min and then incubated with 5 μg/mL of Hoechst 33342 to stain the cell nuclei for 30 min. Images were captured using Olympus IX73 microscope. The percentage of EdU-positive cells in each filed (six random fields were counted in each assay) was defined as the proliferation rate. All the experiments were performed in triplicate.
Colony formation assay
AGS and SGC7901 cells (1×103) were seeded into 6 well plates. After 24 h, cells were treated with Sophoridine (3 μM) at the indicated concentrations for 24 h. Cells were then cultured in fresh medium for another week. Colonies fixed with methanol and stained with 0.05% crystal violet for 30 min. Photographs were acquired and colonies containing more than 50 cells were counted. All the experiments were performed in triplicate.
Immunofluorescence assay
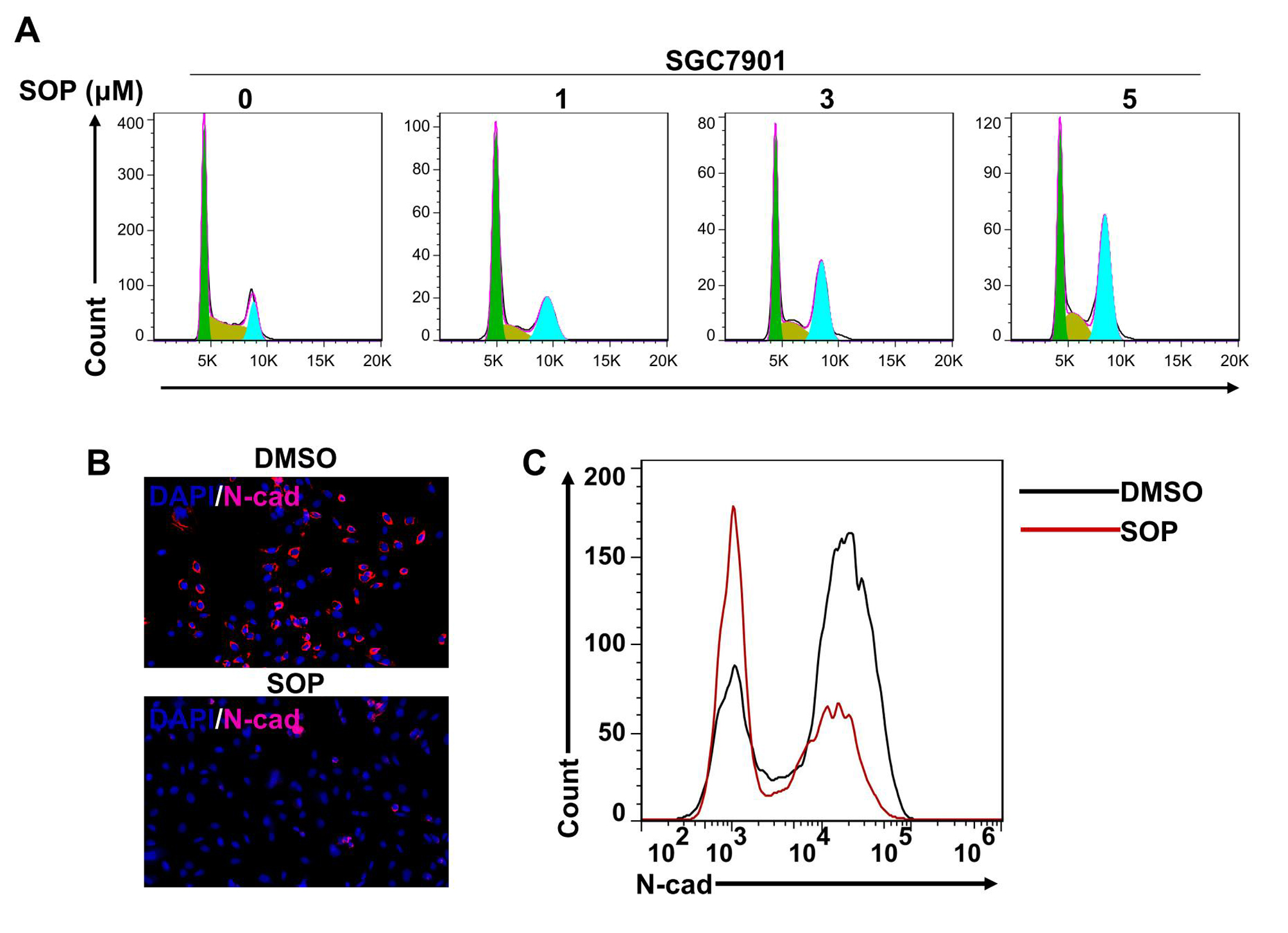
Cells were seeded in 24 well plates and treated with Sophoridine at indicated concentrations for 24 h. The cells were washed in cold PBS and then fixed with 4% paraformaldehyde. Subsequently, cells were blocked with 1% BSA containing 1% goat serum for 30 min. After incubation with mouse monoclonal antibodies to E-cadherin or N-cadherin (Abcam, Shanghai, China) overnight at 4 °C, cells were exposed to Alexa Fluor® 647 labelled goat polyclonal secondary antibody (Abcam) for 1 h at room temperature, and then stained with DAPI. Cells were observed by using Olympus IX73 microscope.
Cell transfection
ESRRG and its non-targeted control (siNC) siRNAs were synthesized from RiboBio (Guangzhou, China). Transfection were performed with Lipofectamine 3000 (Invitrogen) following the manufacturer’s protocol. Selective silencing performance was identified by western blotting.
Preparation of nuclear and cytoplasmic fractions
Nuclear and cytoplasmic extractions were performed using an NE-PERTM Nuclear Cytoplasmic Extraction Reagent kit (Thermofisher, Shanghai, China) according to the manufacturer’s protocol. Briefly, the treated cells (2´106) were washed twice with ice-cold PBS and centrifuged at 500 g for 3 min. The cell pellet was then suspended in 200 μL of cytoplasmic extraction reagent I (CER I) by vertexing. The suspension was subsequently incubated on ice for 10 min followed by the addition of 11 μL of a second cytoplasmic extraction reagent II (CER II), vertexing for 5 s, incubation on ice for 1 min, and centrifuged at 16000 g for 5 min. The supernatant (cytoplasmic fraction) was transferred to a pre-chilled tube. The insoluble pellet fraction, which contains crude nuclei, was resuspended in 100 μL of ice-cold nuclear extraction reagent (NER) by vertexing for 15 s every 10 min over a total period of 40 min, and then centrifuged at 16000 g for 10 min. The resulting supernatant constituted the nuclear extract.
Western blot
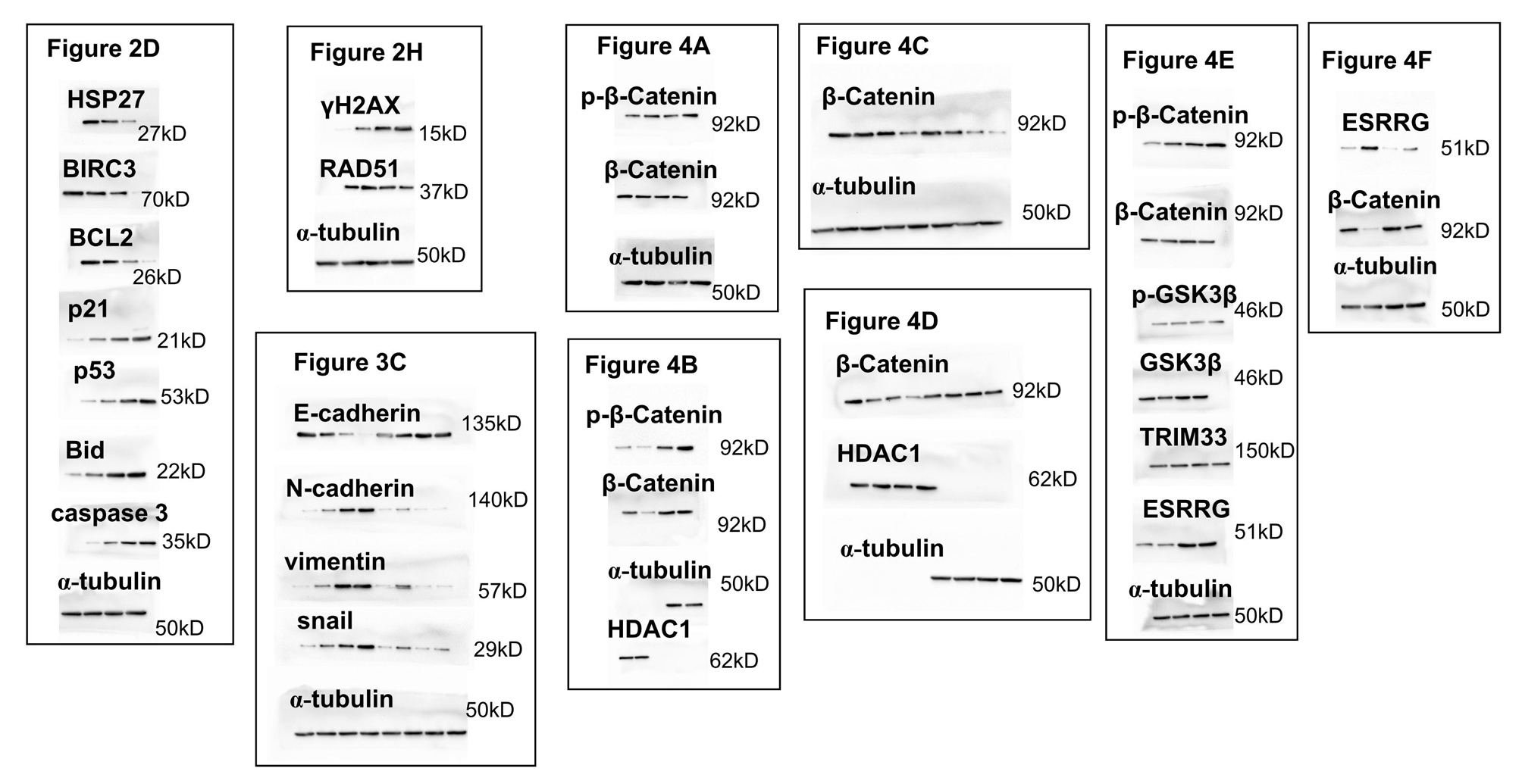
Cells were seeded in six-well plates and treated with indicated conditions. At the indicated time, total proteins from the cells were extracted with RIPA buffer (Beyotime) containing proteinase inhibitor. Protein concentrations were measured by the BCA reagent kit (Beyotime) and then equal amounts of proteins were separated by sodium dodecyl sulfate -polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane (Millipore, USA). The membranes were blocked with 5% non-fat dry milk in TBST buffer for 1 h, and then probed with primary antibodies against HSP27, BIRC3, p53, Bid, pGSK3β, GSK3β, E-cadherin, N-cadherin, Vimentin, snail, TRIM33, ESRRG (Abcam), caspase 3, total and phospho-β-catenin, γH2AX, RAD51, α-tubulin (CST), p21, BCL2, HDAC1 (Santa Cruz) at 4 °C overnight. The membranes were then washed in TBST buffer and incubated with anti-mouse or rabbit horseradish peroxidase-conjugated secondary antibodies. Protein expression were visualized using the enhanced chemiluminescence system (Millipore).
Flow cytometry
For cell cycle analysis, cells were trypsinzed, washed in PBS, fixed in 70% ice-cold ethanol and stored at -20 °C overnight. Samples were then re-suspended in PBS and stained with 50 μg/mL propidium iodide (PI) solution containing 0.2% Triton X-100 and 100 μg/mL DNase-free RNase A for analysis. For apoptosis analysis, cells were harvested and stained using FITC-Annexin V/PI apoptosis detection Kit (BD Biosciences) according to the manufacturer’s instructions. For E-cadherin and N-cadherin expression, cells were trypsinized and washed in cold PBS. Subsequently, cells were blocked with 1% BSA containing 1% goat serum for 15 min and then incubated with primary antibodies to E-cadherin or N-cadherin for 20 min. Next, cells were exposed to Alexa Fluor® 647 labelled goat polyclonal secondary antibody (Abcam) for 1 h at room temperature. Cells were analyzed by using FACSCalibur (Becton Dickinson). Data analysis was performed using FlowJo version 7.6.1 software (TreeStar).
Transwell assay
AGS and SGC7901 cells treated with indicated conditions were resuspended in serum-free RPMI-1640 medium, and 1 × 105 cells were seeded into the upper 24-well chambers (8- µm pore size, Corning Costar). RPMI-1640 medium containing 20% FBS was added to the lower chambers. After 24 h, cells remaining on the upper surface of the membrane were removed with a cotton swab, and the cells that had migrated/invaded into another side of the membrane were fixed with methanol for 15 min. And then, cells were stained with 0.05% crystal violet for 30 min and photographed under Olympus IX73 microscope. The number of migration cells in each filed (six random fields were counted in each assay) was counted from three independent experiments.
Statistical Analysis
Results were expressed as mean ± SD and analyzed by using the Graphpad Prism V.5.00 software (GraphPad Software, CA, USA). Unpaired t-test or one-way ANOVA followed by Neuman-Keuls post-hoc test was used to determine the significance of the difference between groups. P<0.05 was considered statistically significant.
{kind=link}
{kind=link}
{kind=link}