GPATCH2 is a putative repressor of TNF expression
The TNF 3’UTR contains sequences that participate in the post-transcriptional regulation of this gene. With a series of reporter constructs missing one or two of these regulatory elements (Fig. 1A), we have previously shown that deletion of the AU-rich element (ARE) or the new regulatory element (NRE) both increase the basal expression of TNF, and that concomitant loss of both elements increased TNF expression to a very high level (Fig. 1B)10. These results have now been confirmed in vivo6. Since the ARE has been shown to be the target of several RNA-binding proteins, we hypothesised that the function of the NRE is similar and that several RBPs cooperate in vivo to maintain low TNF levels and thereby prevent aberrant inflammation. In order to identify potential RBPs involved in this process, we engineered a CRISPR screen that would mimic the effect of the double deletion by ablating the effect of region 4 when using the Del6 construct and the effect of region 6 when using the Del4 construct.
Cas9-mCherry-expressing HEK293Ts were stably transfected with GFP-Tnf Del4_PGK-Hygro or GFP-Tnf Del6_PGK-Hygro reporter constructs (Fig. 1A) and selected with hygromycin. Single mCherry-Cas9 expressing HEK293T cell clones with moderate GFP reporter expression were selected and expanded (Fig. 1C). Six independent clones were infected with the lentiviral Human GeCKO v2 sgRNA library11 and selected on puromycin. 7 days following infection, GFP-high cells were sorted (Fig. 1D) and put back in culture. After expansion, enriched GFP-high cells were sorted (Fig. 1E) and their DNA was immediately prepared for analysis of their sgRNA content.
In this screen, increased GFP fluorescence following application of the CRISPR library indicated greater reporter mRNA stability, presumably through the loss of a negative regulator of the TNF 3’ UTR. From our reporter screen utilising an NRE deletion construct, high GPATCH2 sgRNA counts were present in two independent GFP-high clones. The presence of a unique GPATCH2 sgRNA in each of these two clones, one targeting exon 3 of GPATCH2 and the other exon 7, suggested a true effect on reporter expression due to specific loss of GPATCH2. Indeed, GPATCH2 was the only candidate that was found more than once in this screen. Since the G-patch domain is found in proteins involved in RNA processing, we decided to proceed immediately with ablating the gene in the mouse, anticipating that loss of GPATCH2 would deregulate Tnf expression and cause an inflammatory phenotype.
Gpatch2 −/− animals develop normally and are healthy
Gpatch2 −/− C57BL/6 mice were generated by CRISPR/Cas9 technology12. sgRNA primers flanking exon 3 were used to excise exon 3 to knock out Gpatch2 (Fig. 2A). Exon 3 was targeted to knock out GPATCH2 as its deletion results in a frame shift with several stop codons generated immediately after exon 2 (Fig. 2B). Founder mice with the expected Gpatch2 gene deletion were identified by PCR and the deletion was confirmed by Sanger sequencing. Mutant founders were then crossed to wild-type C57BL/6 mice to generate true heterozygotes which were then inter-crossed to generate homozygous mutants. Genotypes of offspring were identified with a 3-primer PCR (Fig. 2A).
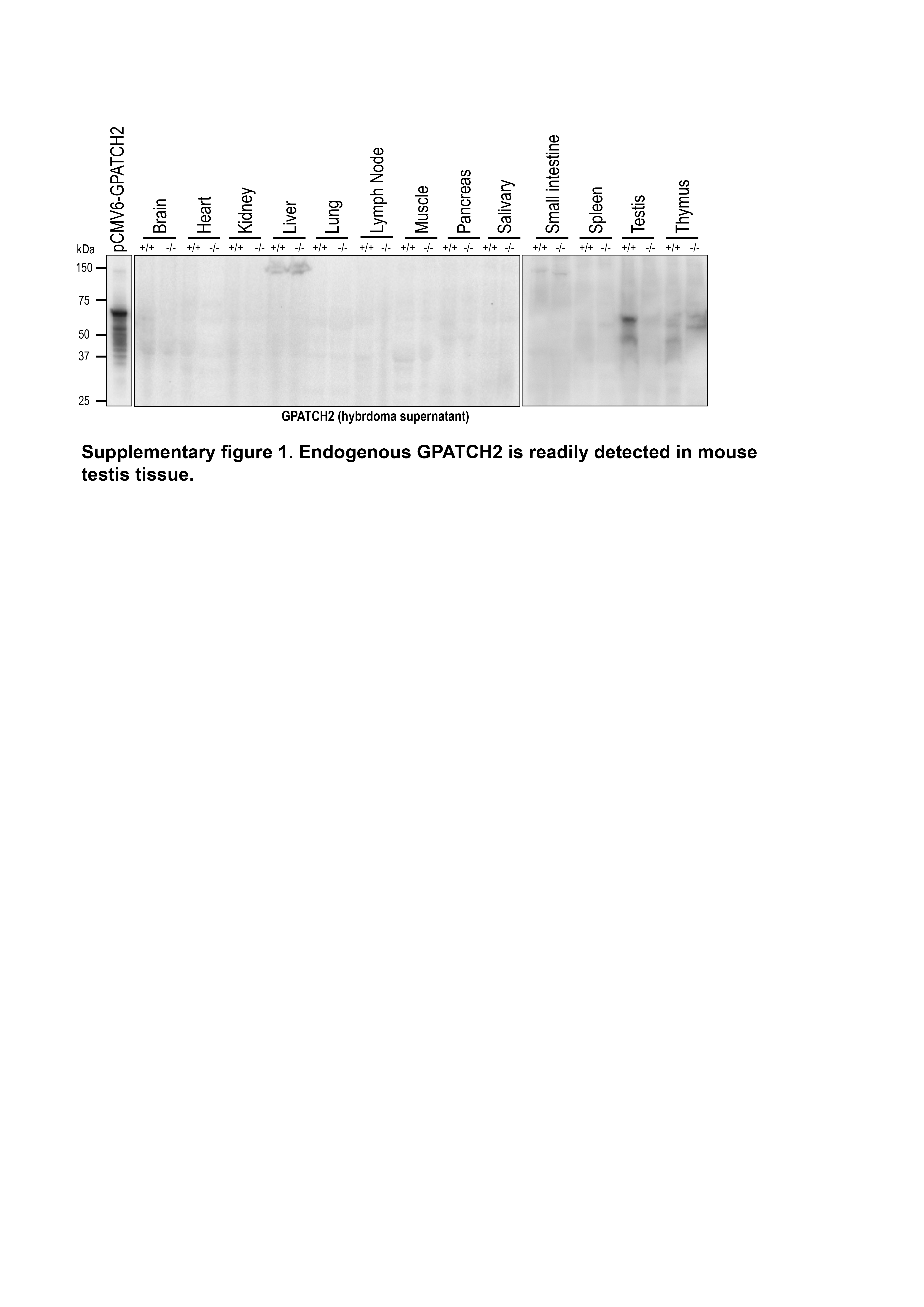
Throughout a two-year observation period, Gpatch2+/− (n > 200) and Gpatch2−/− (n > 200) mice developed and aged normally and mice of the three expected genotypes (+/+, +/- and -/-) from inter-crosses of Gpatch2+/− mice were found at the predicted Mendelian ratios (Fig. 2D). To validate successful CRISPR targeting and Gpatch2 gene knockout, we tested several commercially available antibodies against GPATCH2 without successful detection of endogenous GPATCH2 (data not shown). Thus, we generated a GPATCH2 antibody using a N-terminally truncated recombinant mouse GPATCH2 protein comprised of exons 4–10 as the immunogen. Various organ lysates from wild-type and Gpatch2−/− mice were screened with several hybridoma supernatants for GPATCH2 expression, and a number of hybridoma supernatants were successful in detecting endogenous GPATCH2 protein in the testis (data not shown). With such antibodies we then confirmed successful knockout in Gpatch2−/− mice (Fig. 2C). We cannot exclude the possibility that a N-terminally truncated GPATCH2 protein may exist in Gpatch2−/− mice, however, the G-patch domain contained within exon 10 that is essential for the function of GPATCH proteins was successfully deleted.
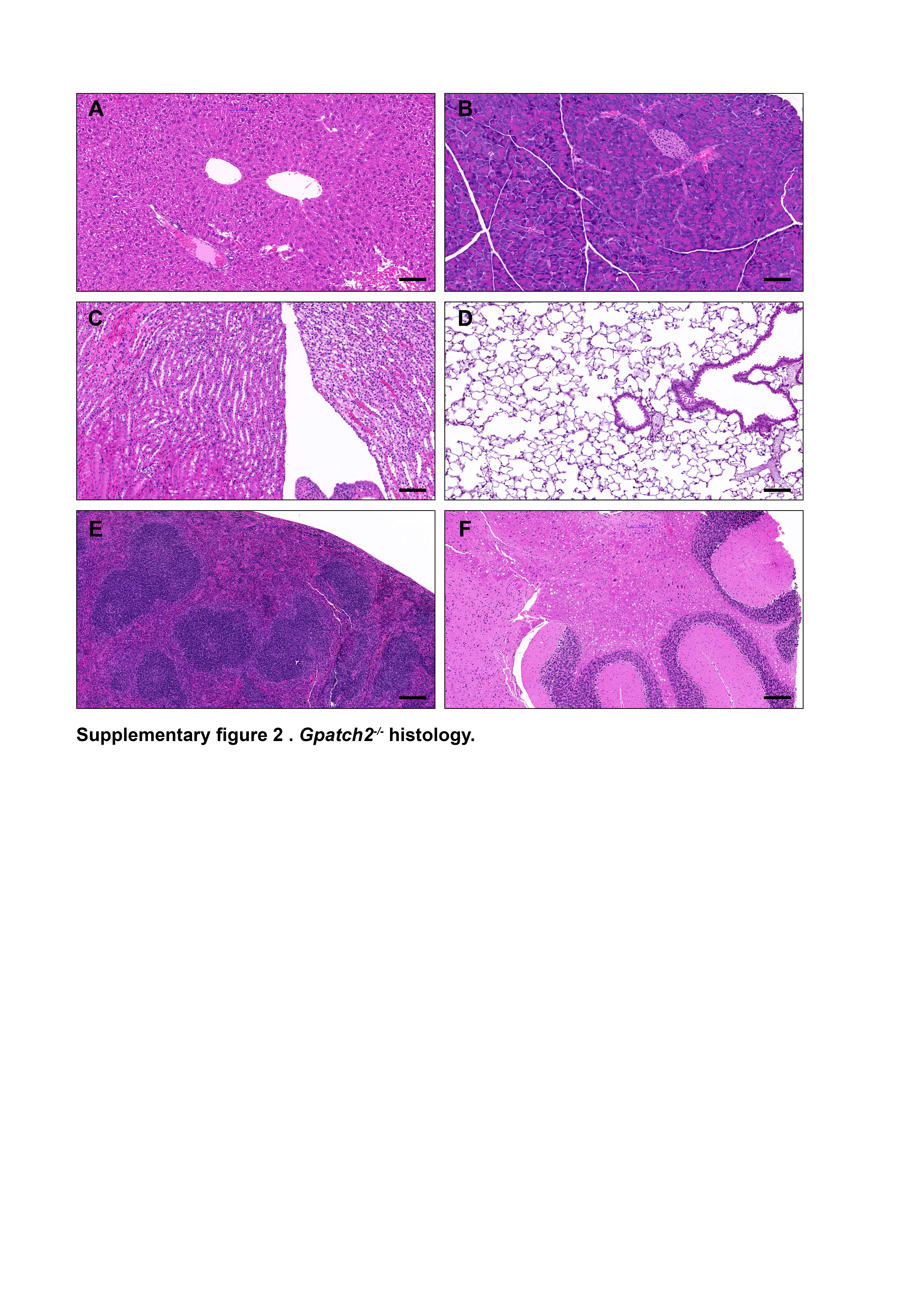
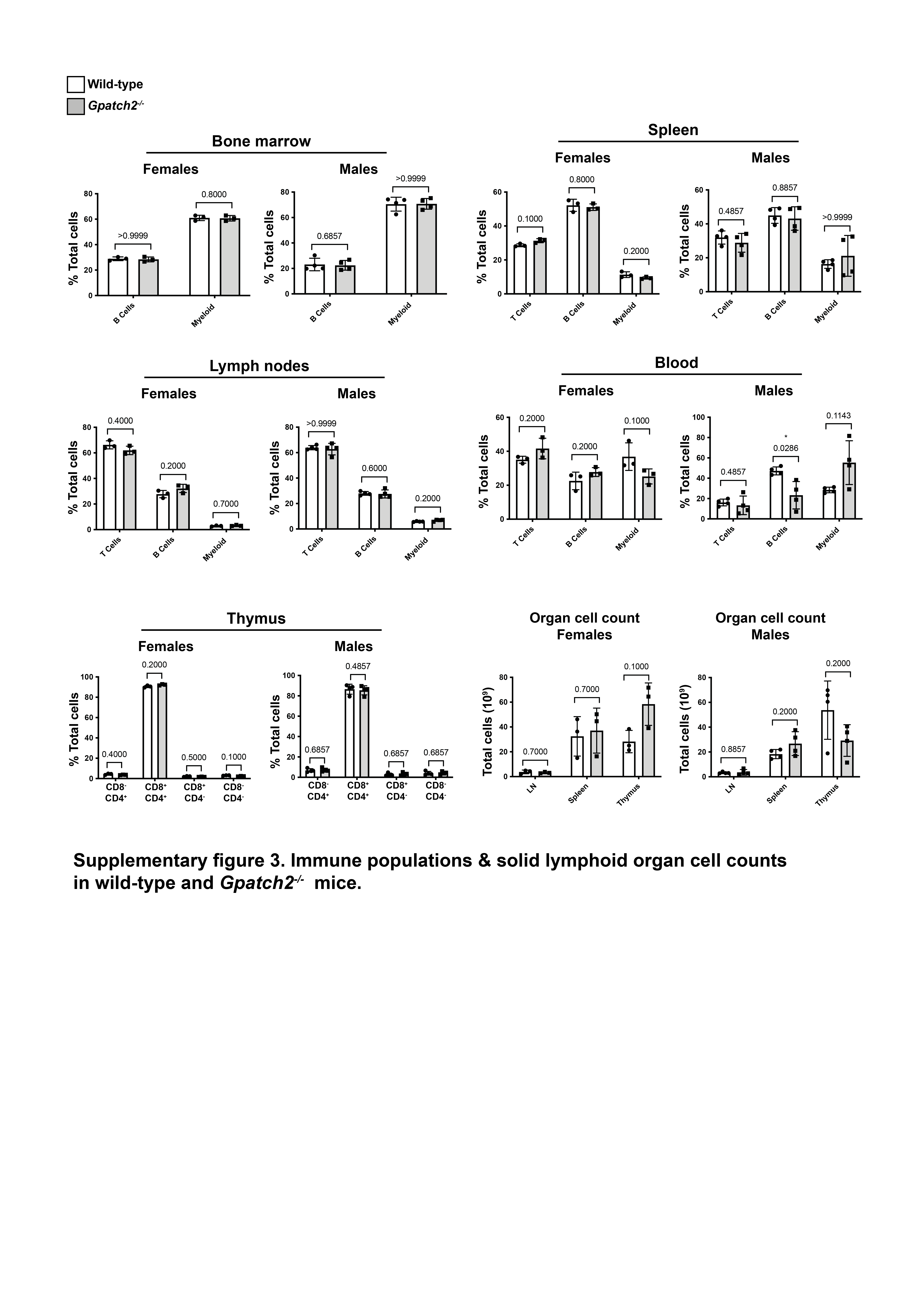
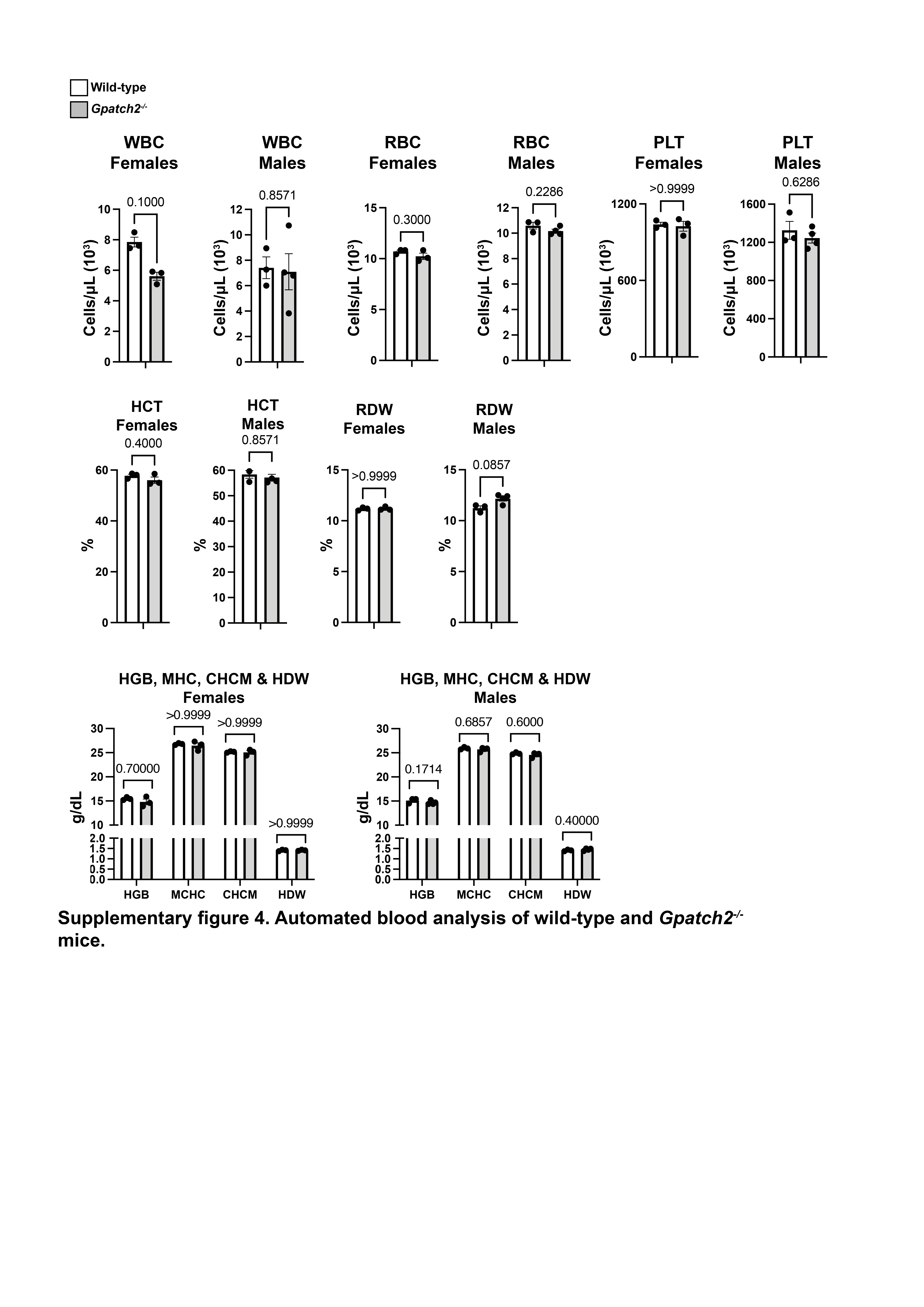
Finally, we screened additional organ lysates for GPATCH2 with our most sensitive hybridoma supernatant, but reliable detection of endogenous GPATCH2 occurred only in the testis (Supplementary Fig. 1). Considering that serum TNF levels remain undetectable in unchallenged Gpatch2−/− mice (Fig. 4C, see pre-LPS injection serum TNF), it is not surprising that Gpatch2 knockout mice did not display an increased propensity for TNF-driven arthritis, inflammatory bowel disease or heart valve disease (Fig. 3A – E), pathologies that are readily observed in a number of our Tnf 3’ UTR mutant mouse strains6, 10. Histological analysis of other tissues from Gpatch2−/− mice also revealed no overt abnormalities (Supplementary Fig. 2). GPATCH2 was first described as a testis antigen and we confirmed expression of GPATCH2 in this tissue (Fig. 2C). However, testis tissue in both young and aged males appeared unaffected by the loss of GPATCH2 (Fig. 3F), and Gpatch2−/− males were perfectly fertile. To screen for alterations to the immune system, lymphoid organ cell counts and cell composition were examined by flow cytometry in male and female Gpatch2−/− mice at 6–10 weeks, but no clear differences were observed in the knockout animals compared to wild-type controls (Supplementary Fig. 3). Other haematological parameters measured by automated blood analysis also yielded results comparable between Gpatch2−/− and wild-type mice (Supplementary Fig. 4).
Loss of GPATCH2 does not affect in vivo response to LPS in mice
Because of the absence of an overt phenotype in Gpatch2−/− mice and no detectable increase in baseline serum TNF levels in Gpatch2-deficient animals (Fig. 4C), we next sought to evaluate whether loss of GPATCH2 may alter TNF production in response to stress stimuli in vivo. Intraperitoneal injection of lipopolysaccharide (LPS) induces a systemic inflammatory response in mice where binding of LPS to the TLR4/MD2 complex activates NF-κB, leading to production of pro-inflammatory cytokines, including secretion of large quantities of TNF from monocytic cells13.
Wild-type and Gpatch2−/− mice were injected with 18 mg/kg body weight LPS14, and all mice showed a similar decrease in body temperature over 3 h post-injection regardless of genotype, and were euthanised at this time due to reaching the ethically-determined endpoint of a 33°C temperature. Blood serum collected post-mortem revealed comparable serum TNF levels between wild-type and Gpatch2−/− mice (Fig. 4A). To slow down the development of symptoms, mice were then injected with 5 mg/kg body weight LPS in order to detect potential differences in response between wild-type and Gpatch2−/− mice. Animals were bled pre-LPS injection to establish baseline serum TNF levels but TNF remained undetectable in the serum of the Gpatch2−/− mice. No difference in symptom severity or serum TNF levels 2 h post-injection were observed at this lower dose of LPS (Figs. 4B & C).
Since macrophages are the main producers of TNF, we also stimulated bone marrow-derived macrophages (BMDMs) from wild-type and Gpatch2−/− mice with LPS in vitro. Induction of Tnf expression was assessed by qPCR, and Tnf mRNA levels were comparable between wild-type and Gpatch2−/− BMDMs following LPS treatment (Fig. 4D). TNF protein levels in culture supernatants remained similar between wild-type and Gpatch2−/− BMDMs throughout LPS treatment and after withdrawal of LPS (Fig. 4E). To confirm Gpatch2 expression in BMDMs, RT-PCR was performed using a forward primer positioned across the exon 2-exon 3 junction to ensure specificity for GPATCH2, where amplification cannot occur in cells from Gpatch2−/− mice where exon 3 is absent. Gpatch2 mRNA was confirmed to be present in wild-type BMDMs by RT-PCR and, as expected, absent in BMDMs from Gpatch2−/− mice (Fig. 4F). Despite apparent expression of Gpatch2 mRNA in BMDMs, our in vivo and in vitro analysis of LPS-induced TNF expression suggested no discernible role for GPATCH2 in the control of Tnf expression in these LPS stimulation models.
GPATCH2 does not affect TNF expression in the SMAC-mimetic-induced skin inflammation model
To further evaluate a potential role for GPATCH2 in the control of TNF expression, we next sought to examine the impact of loss of GPATCH2 in the context of TNF-dependent skin inflammation. SMAC-mimetic compounds (SMs) antagonise inhibitor of apoptosis proteins (IAPs), predisposing to cell death. Subcutaneous injection of the SMAC-mimetic Compound A (CompA)15 depletes IAPs in the skin surrounding the site of injection, and a lesion is formed as a result of a localised acute inflammatory reaction within the epidermis16. This inflammatory reaction in the skin is TNF-dependent, as Tnfr1−/− mice do not develop such a reaction following CompA injection16.
Gpatch2 −/− and control wt mice were injected subcutaneously with CompA on one side of the flank and injected with CompA on the opposite flank two days later for the 1 and 3 day timepoints. Unaffected skin was collected from the ventral thoracic region of each mouse as untreated control skin. Mice were euthanised three days after the first CompA injection and hair removed with a depilatory cream before photography of lesions, as previously described16. Day 3 lesions were scored for redness and oedema, as well as the Nikolsky sign, an indicator of epidermal detachment17. No clear macroscopic differences were observed between the inflammatory lesions of Gpatch2−/− animals and wild-type controls (Fig. 5A & 5B). H&E staining of lesions revealed comparable epidermal and dermal injury between wild-type and Gpatch2−/− animals, while staining for activated (i.e. cleaved) caspase-3 (CC3) revealed no difference in the quantity or pattern of apoptotic cell death at both 1 day or 3 days post SM-injection (Fig. 5C). Staining for the nuclear protein Ki67 demonstrated comparable cell proliferation in lesions from wild-type and Gpatch2−/− mice (Fig. 5C). Day 1 lesion centres and Day 3 lesion edges are shown in Fig. 5, as differences in the response to CompA injection are most apparent at the site of injection, or lesion centre, on day 1 where considerable keratinocyte death occurs and at the lesion edges on day 3 where wound healing is occurring. Loss of GPATCH2 had no apparent impact on the expression of the inflammatory cytokines TNF, IL-6 and MCP-1 in skin lesions at either 1 day or 3 days post-injection (Fig. 5D). Collectively, these results show that Gpatch2−/− mice do not display a heightened response to SM injection, nor elevated TNF in SM-induced lesions, This suggests that GPATCH2 is not essential to regulate Tnf expression in the SM injection model.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}