Characterization of PGL, EUO, and Biopolyols
The characteristics OH, acid, and functionality values of the EUO, PGL and biopolyols are determined according to [37-42] and shown in table 3.
Table 3 Characteristics of the PGL, EUO, and biopolyols
|
|
PGL
|
biopolyol-1
|
biopolyol-2
|
biopolyol-3
|
biopolyol-4
|
|
OH value
(mg KOH/g)
|
270 ± 30
|
160 ± 20
|
140 ± 20
|
128 ± 20
|
360 ± 40
|
|
Functionality
|
-
|
6.8
|
5.95
|
5.4
|
15.3
|
|
Acid value
(mg KOH/g)
|
1.97
|
2.8
|
1.4
|
2.24
|
9.8
|
|
Functionality
|
-
|
0.22
|
0.13
|
0.78
|
2
|
|
Viscosity average
(mPa.s)
|
689
|
54
|
37
|
831
|
2519
|
|
Content of glycerol[1] in CG was 34%
|
The 1HNMR spectra of UCO, PGL, and BD are presented in Fig. 6-8-a. The spectrum of UCO illustrates the most important peaks such as terminal -CH3, -(CH2)- Chain, β-CH2, -CH2-CH2-CH=, α-CH2, =CH-CH2-CH=, -CH-CH2-O(CO)-R, -CH2-CH(CO-R)-CH2-, and -CH=CH- at 0.80 ppm, 1.20 ppm, 1.60 ppm, 2.00 ppm, 2.25 ppm, 2.75 ppm, 4.00-4.5 ppm, 5.25 ppm, and 5.30-5.50 ppm, respectively. Clear differences were observed in both the PGL and BD spectra. For instance, multiple peaks at (a) and one peak at (c) ppm relate to -CH-CH2-O-H and -CH2-CH(OH)-CH2- in the PGL spectrum. More, what distinguishes the spectrum of BD is the strong peak at 3.20 ppm which corresponds to -(CH2)-, and the peak at 4.20 ppm which corresponds to -OCH3. When comparing PGL 1HNMR spectrum with 1HNMR spectrum of commercial glycerin, Fig. 9, peaks (a, b, and c) are identical and represent the protons of a single molecule of glycerol or linear polyglycerol. The (d) peak may represent the proton attached to the carbon atom that is part of R. The high area of the (d) peak indicates the presence of a significant amount of branching structures.
The PGL & BD were detected in UCO vs. PGL & BD FTIR spectra, Fig. 6-8-b. The intensity of peak in the UCO spectrum at 3001 cm-1 corresponded to C-H of alkene, -CH3 asymmetrical stretching at 2930 cm-1 and symmetrical bending at 1370 cm-1, -CH2 asymmetrical stretching at 2850 cm-1, and scissoring at 1455 cm-1. In addition, a strong peek at 1740 cm-1 because of C=O ester stretching vibrations, a small peak at 1655 cm-1 relates to C=O free fatty acids, bending of C-O bands at 1250, 1155, 1078 cm-1, and -(CH2)n- rocking at 714 cm-1. As for the BD spectrum, a broad hydroxyl band appeared at 3383 cm-1, which may be due to the presence of moisture. A peak at 1640 cm-1 corresponded to C=O, a strong peek at 1018 cm-1, and peaks appeared at 1466 cm−1 related to C-O and CO-O-CH3 which indicated that the methyl esters are formed. The PGL FTIR spectrum was obtained in the Fig. 8-b. There is a -CH asymmetrical stretching peak at 2930 cm-1 and a -(COH) small peak at 1450 cm-1. There is a strong peak in 1641 cm-1 may be due to the C=O bond indicating the presence of mono- and diacyl glycerides. Hence, a broad hydroxyl band of glycerol appeared at 3400 cm-1.
The 1HNMR spectra illustrated some changes in the signals. The emergence of new areas in the range of 2.80-3.00 ppm belonging to the hydrogens attached to the oxirane ring in the spectrum of EUO coincided with the partial disappearance of some others in the range of 5.30 - 5.50 ppm belonging to the olefinic hydrogens in the spectrum of UCO. On the other hand, the same areas of 2.80-3.00 ppm disappeared completely to show new ones in the range of 3.40 – 4.40 ppm in the biopolyols' spectrum. In biopolyol-2, the signal at 4.50 corresponded to hydrogens of diethylene glycol, and the signal at 8.15 ppm related to hydrogens of the aromatic cycle of PET. In contrast, weak signals at 6.25-8.25 ppm are derived from the hydrogens of aromatic cycle isocyanate in PU. Likewise, peaks at 6.50-7.50 ppm in the biopolyol-4 are caused by the aromatic cycle of BPA, Fig. 10-14-a.
The EUO and biopolyols were detected in FTIR spectra Fig. 10-14-b. The intensity of peak in UCO spectrum at 3001 cm-1 corresponded to C-H of alkene was decreased and disappeared in the EUO spectrum, accompanied by appearing a peak at 840 cm-1 corresponding to oxirane ring. Generally, there are participated peaks such as a broad hydroxyl band appeared at 3400 cm-1 and C=O stretching vibrations at 1700 cm-1 in all novel biopolyols' spectra. More, the peek corresponded to the epoxide cycle was faded. The absorption bands at 1000-1200 cm-1 correspond to the aliphatic ether group of diethylene glycol. The bending vibrations of the methylene groups in the biopolyol chain are also consistent at 1350-1450 cm-1. The CH bonds in the aliphatic carbon are stretched at 2900-2800 cm-1. Some additional signals appeared such as 1500 cm-1 due to aromatic C=C stretching in biopolyol-2 and biopolyol-4, 1500 cm-1 and 1600 cm-1 related to amine groups in biopolyol-3. [43-46].
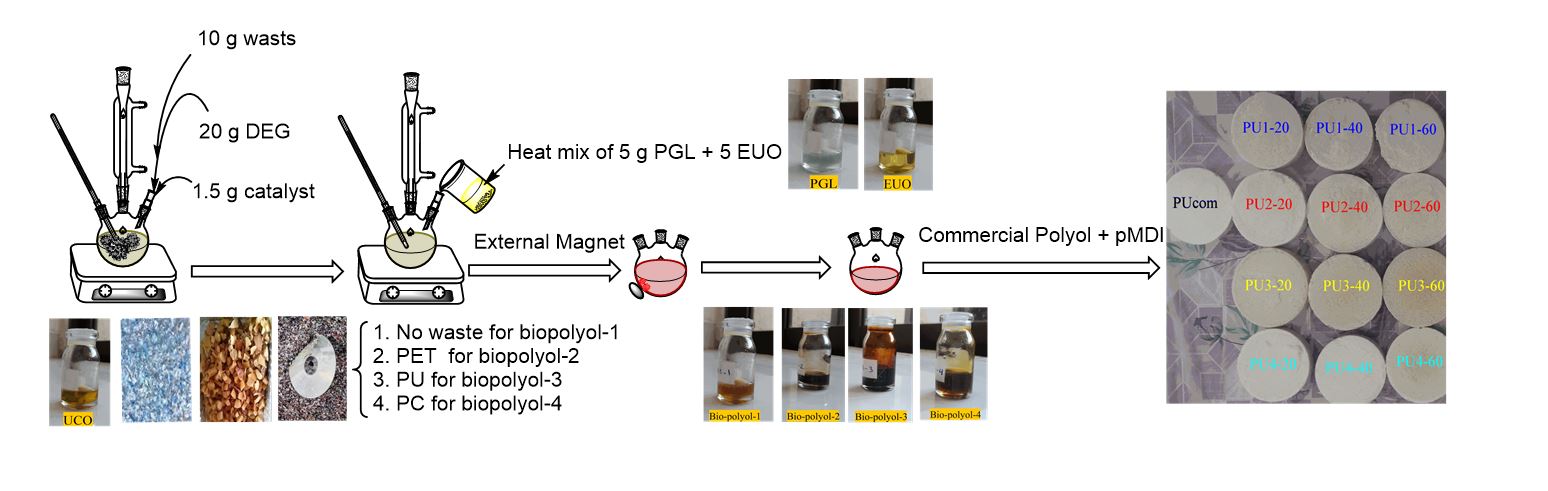
Characterization of Polyurethane Rigid Foams
The foam during formation usually goes through steps where the mixture turns first into a cream and then into a gel, accompanied by the rise of the mixture and the formation of foam. The stirring, cream, gel, and rise times were determined for foams and their composites. The results show that the blend of biopolyol-4 takes more time in rise operation. While the values are close for other biopolyols. The reason can be attributed to the type of polyol where the reaction with biopolyol-4 takes more time than other types because of high viscosity. The apparent density of foams directly depends on the viscosity of the mixture of components as high viscosity inhibits cell growth. On the other hand, viscosity is related to the degree of compatibility between these components. It can be seen that the apparent density gradually increases with the increase in the biopolyol content for all biopolyols, table 4, [47-50].
The thermogravimetric analysis is one of the useful methods used for analyzing the thermal degradation of polymers, especially polyurethane rigid foams and their composites. The curves of weight loss (TG) and weight loss derivatives (DTG) in the air atmosphere present in Fig. 15, while the results are summarized in table 4. Generally, changes on polyurethane foams occur after a temperature of 100 °C. Initially, the hydrogen bonds between the urethane groups and the oxygen atoms of the soft parts and the allophanates and urea bonds are broken within the range 100 – 180 °C. The main urethane bonds break after 200 °C degrees to destroy the solid part at 250 °C. Heat breaks urethane bonds with the type of substituents (aliphatic or aromatic) located on both terminals of the urethane group where the urethane groups with aliphatic substituents are the most resistant to thermal cracking. This is followed by the decomposition of the soft parts and then the ester groups in the soft parts. The curves showed two main steps, the first between 180 °C and 430 °C and the second between 430 °C and 700 °C, corresponding to 45% and 55% mass loss, respectively. The first step refers to the degradation of the hard segments including many kinds of bonds and perhaps the most important of which is urethane bonds, which start to break down first, and ester bonds in the soft parts, which start to break down starting from 380 °C. The second step may refer to the decomposition of flexible segments, aromatic compounds or thermolysis of organic residues. Each has a maximum degradation rates Tmax2 = 346 - 357 °C and Tmax3 = 570 - 598 °C for based-biopolyol-1 PURFs, Tmax2 = 346 - 354 °C and Tmax3 = 584 - 586 °C for based-biopolyol-2 PURFs, Tmax2 = 317 - 356 °C and Tmax3 = 584 - 586 °C for based-bio-polyol-3 PURFs, and Tmax2 = 286 - 373 °C and Tmax3 = 577 - 610 °C for based-biopolyol-4 PURFs.
The introduction of biopolyols significantly affected the temperature at the beginning of the decomposition T5% compared with the comparative sample, as it caused a decrease in the temperature for all based-biopolyols PURFs at all ratios except for based-biopolyols-3 & 4 PURFs at ratio 20% PU3-20 and PU4-20, which have increased. The decrease was notable for all foams with ratio 60% PU1-60, PU2-60, PU3-60, and PU4-60. The introduction of biopolyols-1, 2, 3, and 4 at ratio 60% for the fabricated foams led to some changes in the foam structure and observation of an additional step in the thermal decomposition corresponding to the degradation of the rigid segments Tmax1. As the biopolyol content increases 40%, 60%, the degree of phase separation in polyurethane foam increases. Probably, causes the pyrolysis pathway of the foam to be changed to form a three-phase state which results in a 10% and 20% reduction in weight loss of PU1-60, PU2-60, PU4-40 & 60, and PU3-60, respectively. The results of the analysis, when compared with the reference sample, indicate sometimes higher or equal thermal stability for foams PU1-40, PU2-40, and PU3-20, and sometimes less for other foams, as evident from the values of the table at the 50% mass loss temperatures. The values of Vmax from the curves for the first decomposition steps show that the decomposition speed of the reference sample is slower than the rest of the samples, but at the middle and last steps, it shows a different behavior as it becomes faster except for foams PU2-60, PU4-60. The percentage of combustion residues at 800 °C has no significant effect for foams after the introduction of biopolyols [51-56].
Table 4 Characterization of foaming process, foam properties, and summary of TGA results of foams
|
|
X
|
Stirring time (sec)
|
Cream time (sec)
|
Gel time (sec)
|
Rise time (sec)
|
Apparent density[2] (g/cm3)
|
T5%,
(°C)
|
T50%, (°C)
|
Tmax1, (°C)
|
Tmax2, (°C)
|
Tmax3, (°C)
|
Residue (%)
|
|
PUx-20
|
1
|
6
|
11
|
22
|
35
|
0.094
|
236
|
441
|
-
|
357
|
571
|
0
|
|
2
|
6
|
11
|
24
|
38
|
0.042
|
219
|
483
|
-
|
354
|
585
|
0
|
|
3
|
6
|
11
|
24
|
38
|
0.040
|
275
|
514
|
-
|
356
|
600
|
0
|
|
4
|
6
|
12
|
30
|
60
|
0.061
|
265
|
449
|
-
|
373
|
577
|
0
|
|
PUx-40
|
1
|
6
|
10
|
23
|
36
|
0.095
|
229
|
507
|
-
|
436
|
598
|
0
|
|
2
|
6
|
11
|
23
|
38
|
0.050
|
227
|
501
|
-
|
353
|
584
|
0
|
|
3
|
6
|
11
|
24
|
38
|
0.065
|
237
|
469
|
-
|
345
|
585
|
0
|
|
4
|
6
|
12
|
30
|
61
|
0.066
|
90
|
465
|
125
|
337
|
603
|
0
|
|
PUx-60
|
1
|
6
|
12
|
24
|
35
|
0.096
|
98
|
482
|
110
|
356
|
581
|
0
|
|
2
|
6
|
12
|
24
|
40
|
0.064
|
172
|
494
|
190
|
346
|
586
|
0
|
|
3
|
6
|
11
|
24
|
38
|
0.068
|
94
|
322
|
122
|
317
|
583
|
0
|
|
4
|
6
|
12
|
31
|
64
|
0.075
|
58
|
547
|
89
|
286
|
610
|
0
|
|
PUcom=
|
|
5
|
10
|
23
|
37
|
0.047
|
238
|
501
|
-
|
354
|
584
|
0
|
The changes which occurred in the morphology of cellular structures of foams because of blending of the reference sample with biopolyols which was depicted by SEM, Fig. 16-20. The different behavior can be attributed to the different reactions of biopolyols according to different weight ratio as it is considered a hydrophilic and hydrophobic functional groups container material, which affects the morphological and structure of the pores including cracks in the walls, collapse, and deform at the largest weight percentage. The cells of most of the images show oval shapes and wide range of sizes. More, comparison sample PUcom image shows an average diameter 287 ± 140 µm.
The introduction of biopolyol-1 by weight ratios of 20, 40, and 60% for samples PU1-20, PU1-40, and PU1-60 has led to a gradual increase in the average cells' diameter to be 353 ± 103, 339 ± 103, and 313 ± 92 μm, respectively. The sample of PU1-60 has some deformation, Fig. 17-d. The addition of biopolyol-2 for samples PU2-20, PU2-40, and PU2-60 has led to a significant increase in the average pore diameter comparing with PUcom, the increase was gradually by 383 ± 104, 393 ± 120, and 436 ± 220 µm, respectively. The average diameter of the foam cells has increased significantly upon introduction of biopolyol-3 from 287 ± 140 µm for PUcom to 495 ± 166 µm, 438 ± 147 μm, and 460 ± 219 for 20%, 40%, and 60%, respectively. The mean values of cell diameters increased slightly when biopolyol-4 was added to the comparative sample and ranged between 285 and 334 µm. Nevertheless, it can be seen that the cell density of the blended foams is higher than that of the comparison sample. This can be attributed to the presence of a very small percentage of bubbles or large vacuoles resulting from the fusion of some cells with each other, which led to an increase in the standard deviation of cell diameter in the two samples PU4-20. This happened in the sample PU4-60 but this was accompanied by the collapse and crash of the bubble and this explains the presence of distortions in the structure [57-61].
Dynamic mechanical thermo-analysis (DMTA) is used to study the response of viscoelastic materials to periodic deformation as a function of temperature change. The figures, Fig. 21,22, represents the loss factor (tan δ), the storage modulus (E'), and the loss modulus (E″) of the prepared polyurethane rigid foams. Samples based on the blended biopolyol-1 with commercial polyol show a gradual decrease in the storage modulus with increasing temperature. At 25 °C, a change was observed in the storage factor values for the ratios 20%, 40%, and 60% compared to the reference sample, as it increased for the last mixture other than the first and second blends. The temperature dependence curve of the loss factor shows that the peak value of the loss factor increases with the increase in the content of biopolyol-1, that is, 60%, while at the two percentages 20% and 40%, the values were lower compared to the reference sample. The glass transition temperature was taken from the tan δmax peaks and the values were 209, 215, 197, and 163 °C for samples PUcom, PU1-20, PU1-40, and PU1-60, respectively. It is possible that the addition of a small percentage of biopolyol-1 resulted in better localization of the hydroxyl groups within the polymer matrix to become more available than in the comparison sample. This resulted in more cross-linking points in order to form hydrogen bonds with the isocyanate groups and thus would impede the movement of the polymeric chains or the increase in the solid section and the value of the glass transition. This confirms that the peak heights of the samples PUcom and PU1-20 are equal, given that the height of tan δmax represents the entanglement density. All samples show only one peak, indicating the homogeneous nature of polyurethane foams. The introduction of biopolyol-2 on the comparison polyol at the weight ratios of 20% and 40% reduced the storage modulus E' at 25°C, while no effect occurred at 60%. Also, for the loss modulus E", the peak of PU2-60 was the highest. This behavior is similar to that of biopolyol-1. The glass transition temperatures of the foams PU2-20 and PU2-40 were almost slightly higher than the blank sample except for the foam PU2-60 which was lower and reached 167 °C. The storage modulus decreased at 25 °C at all ratios when biopolyol-3 was introduced to the control polyol. The minimum foam value was at 20 wt%. For the loss modulus, the peak of the PU3-60 foam was the highest. There was a clear difference in the glass transition temperature values for the foams, but two peaks were observed for the PU3-60 foam. This can be attributed to the fact that the presence of a higher proportion of biopolyols resulted in a lack of network integrity due to incompatibility. The storage factor increased at 25 °C for PU4-40 foam when the biopolyol-4 was introduced on the control polyol while the remaining two foams were lower than the control sample. In terms of loss modulus, PU4-40 foam had the highest peak. The glass transition temperature values for the foams gradually decreased in the tan δ diagram [62-67].
[1] The content of glycerol in CG was determined by the method [37]
[2] The densities of foams were calculated by using the formula described in DIN 53479 [50]
{kind=link}