Chemicals
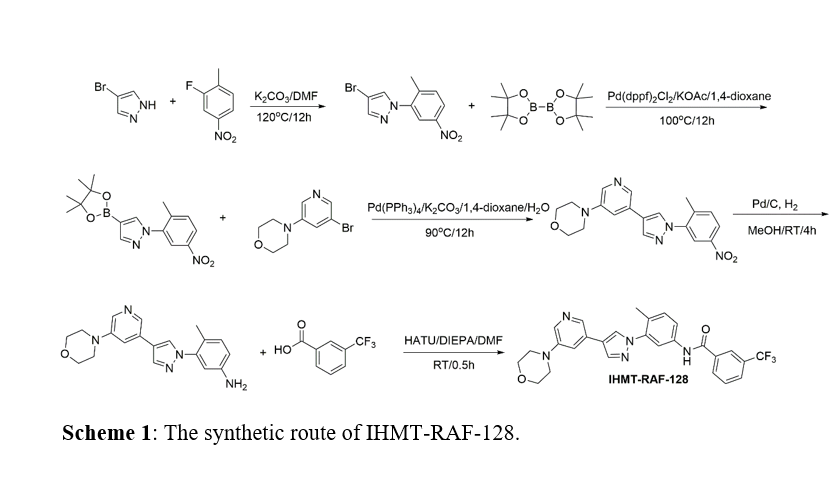
IHMT-RAF-128 was in-house synthesized following the route shown in Scheme 1; Vemurafenib (PLX4032), LY3009120, Encorafenib, Adagrasib, Sotorasib, and RAF709 were purchased from MedChemExpress (Shanghai, P.R.China).
Antibodies and immunoblotting
Phospho-MEK1/2 (Ser217/221) antibody (#9154), MEK1/2 antibody (#9122), Phosphop-ERK1/2 (Thr202/Tyr204) (197G2) antibody (#4377), ERK1/2 (137F5) antibody (#4695), GAPDH (D16H11) XP antibody (#5174), PARP (46D11) antibody (#9532), and caspase-3 (8G10) antibody (#9665) were obtained from Cell Signaling Technology (Danvers, MA, USA).
BaF3 isogenic cell line generation
The functional BaF3 cell lines were constructed as described before[23]. In short, wild-type and mutant BRAF and RAS genes were cloned into the pMSCVpuro retroviral vector and co-transfected with two helper plasmids for virus production in HEK-293T cells. Culture supernatant containing virus particles was used to infect BaF3 cells, and stable BRAF or RAS-overexpression of BaF3 cells were then obtained by puromycin selection and IL-3 withdrawal.
Molecular modeling
The crystal structure of BRAF was obtained from Protein Data Bank (PDB ID: 4G9R). Water molecules and original ligand were manually removed using software[24]. CRAF structure of DFG-out type was modeled using MODELLER[25] based on the template of BRAF. The docking jobs were carried out by Autodock (version 4.2.6)[26]. The prepare_ligand4.py and prepare_recptor4.py scripts from AutoDockTools 1.5.6 were used to prepare the initial files including additions of charges and hydrogen atoms. A grid box of 60*60*60 with a spacing of 0.375Å was set to enclose the whole binding site. The Lamarckian genetic (LGA) was adopted to search the best binding poses. The specific docking settings were as follow: trials of 100 dockings, 300 individuals per population with a crossover rate of 0.8 and the local search rate was set to 0.06. Other parameters were set as default during the docking.
Cell lines and cell culture
H358, Capan-2, KYSE-30, PF382, ASH-3, H2087, BEAS-2B, MCF-10A, A375, HCT116, BxPC3, ASPC-1, HT29, and SK-Mel-28 cell lines were bought from ATCC (American Type Culture Collection). SK-GT-4, CAL62, MDA-MB-231, NALM-6, CAL-12T, 8305C, HEL, CHO cell lines were bought from Cobioer (Nanjing, China). CAL62, MDA-MB-231, CAL-12T, A375, HT29 and HCT116 cells were cultured in DMEM (Corning, USA) with 10% FBS; Capan-2 cells were cultured in McCoy's 5A (GIBCO, USA) with 10% FBS; KYSE-30 were cultured in RPMI1640: Ham’s F-12 (1:1), 2mM L-Glutamine, and 10% FBS; SK-GT-4, H358, PF382, NALM-6, H2087, HEL, BxPC3 and ASPC-1 cells were cultured in RPMI1640 with 10% FBS; ASH-3 cells were cultured in DMEM: RPMI1640 (1:1) with 10%FBS; 8305C, SK-Mel-28 cells were cultured in MEM (Corning, USA) with 10% FBS, NEAA, 1 mM sodium pyruvate, 2mM L-Glut. CHO cells were cultured in F12K (Corning, USA) with 10% FBS. BEAS-2B cells were cultured in BEGM (Lonza, USA) with 10% FBS; MCF-10A were cultured in MEGM (Lonza, USA) with 10% FBS. All cells were cultured in an incubator at 37℃and 5% CO2.
Biochemical assay of kinase activity
The in vitro enzymatic assay was provided by the Beijing ICE Bioscience Company (Beijing, China). Briefly, BRAF (0.2ng/µL), CRAF (0.13ng/µL), and BRAF-V600E (0.1ng/µL) proteins were incubated with IHMT-RAF-128 (20-10000 nM), MEK1 substrates (BRAF: 2.5 mg/mL, CRAF: 7.1 mg/mL, BRAF-V600E: 2.5 mg/mL), and ATP (BRAF: 25 µM, CRAF: 10 µM, BRAF-V600E: 30 µM). The reactions were initiated by adding ATP to each test tube to and continued at room temperature for 1h. ADP-GLO reagent was then added to each well to terminate the reaction, and the remaining ATP was consumed for 40 minutes. Finally, the kinase detection reagent was added to the wells and incubated for 30 minutes to generate luminescence signals, which were then analyzed on microplate reader (Envision, PE, USA) and Prism 8.0 (GraphPad software) to calculate compound potencies.
Western blot analysis
A375, SK-Mel-28, ASPC-1, and HCT116 cells were treated with serially diluted IHMT-RAF-128, RAF709, and LY3009120 for 2h. Cells were then collected by centrifugation and lysed with RIPA buffer (Beyotime, China). The cell lysates were separated by 10% SDS-PAGE electrophoresis and blotted with antibodies after membrane transfer and blocking. At last, the ECL Western blot detection kit (Millipore) was used to examine the effects by drug treatment.
Cell cycle analysis
A375, SK-Mel-28, ASPC-1, and HCT116 cells were treated with serially diluted IHMT-RAF-128, RAF709, and LY3009120. 70% ethanol and PI/RNase staining buffer (BD, USA) were used to fix and stain cells, which were then analyzed on FACSCalibur™ flow cytometer and ModiFit software to test the effects of drug treatment on cell cycle.
Apoptosis detection
A375, SK-Mel-28, ASPC-1, and HCT116 cells were treated with continuously diluted IHMT-RAF-128, RAF709, and LY3009120 for 48h and 72h. The cells were then collected and measured by Western blot assays using PARP, cleaved-PARP, Caspase3, cleaved-Caspase3, and GAPDH antibodies to detect the effect of compounds on apoptosis.
Mouse xenograft study
Nude mice (5 weeks) were purchased from Vital River Laboratories (Beijing, China). The mouse xenograft studies complied with the animal ethics standards of the Hefei Institute of Physical Science, Chinese Academy of Sciences. Briefly, ten million SK-Mel-28, ASPC-1, and BxPC3 cells were first mixed 1:1 with Matrigel gel and injected into the right subcutaneous tissue of nude mice. When tumor sizes reached around 150 mm3, animals were randomly divided into groups of 4–5 mice each. IHMT-RAF-128 and LY3009120 were orally administered daily. The body weights and tumor sizes of the mice were measured daily. The formula (width2 × length /2) was used to calculate the tumor volumes. Tumor growth inhibition (TGI) was calculated according to the formula: (Wvehicle-Wtest) /Wvehicle × 100%, in which W is the tumor weight.
{kind=link}