This study addresses the molecular mechanisms by which apo- and holo-Tf regulate iron release at the BBB. More specifically, this study demonstrates that apo- and holo-Tf differentially interact with Heph and Fpn. Through its interaction, holo-Tf reduces Fpn protein levels, and this is through Fpn’s established degradation pathway as shown when Fpn degradation is inhibited. Holo-Tf directly interacts with Fpn as shown by orthogonal techniques. Furthermore, when incubated together, hepcidin can interrupt this interaction at high levels that correspond with inflammation or high systemic iron levels, but not at levels that correspond with baseline levels. Hepcidin’s interruption is likely due to its ability to internalize Fpn faster than holo-Tf and not due to direct competition for the same binding site, as we additionally demonstrate herein. On the other hand, hepcidin does not interrupt the interaction between apo-Tf and Heph. These findings offer a glimpse at the mechanism of free iron release into the brain, a crucial process for neurological health.
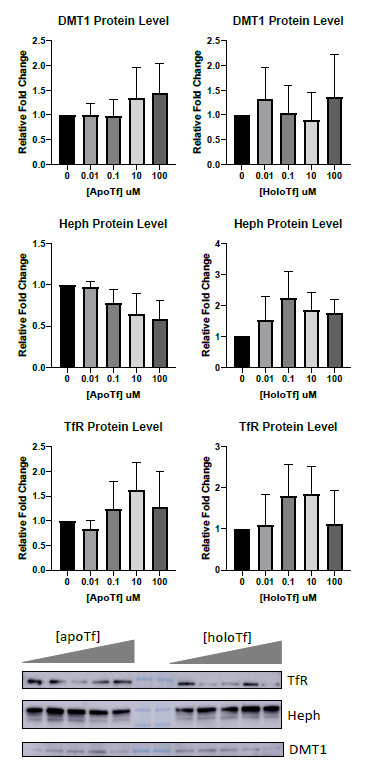
Fpn is the only known iron exporter, thus control of membrane Fpn is control of free iron release. The internalization and subsequent degradation of Fpn has been extensively studied in the context of hepcidin12,15,24,27. Briefly, once hepcidin binds to Fpn, it triggers the ubiquitination of the Fpn, thus signaling for its internalization and lysosomal degradation. Simpson et al. showed that by incubating BRECs with 12.5 µM holo-Tf, the levels of Fpn decreased6. Here we have replicated those findings in iPSC-derived ECs but at a physiological level; transferrin is found in CSF at about 2 mg/dL, or 0.25 µM25. We demonstrate that a basal incubation of as low at 0.1 µM holo-Tf results in a 50% decrease of membrane Fpn. These data provide a mechanistic explanation for why holo-Tf suppresses iron release from ECs. What’s more, other iron-related proteins, such as Heph, DMT1, and TfR, are unchanged with basal holo-Tf exposure. Interestingly, even when exposed to high amounts of holo-Tf, the levels of Fpn do not decrease beyond 50%, suggesting there is a plateaued effect of holo-Tf within the 8-hour experimental time window. The holo-Tf-mediated internalization of Fpn is blocked when the ubiquitination of Fpn is inhibited, suggesting that holo-Tf exerts its effect through the established degradation pathway, similar to hepcidin.
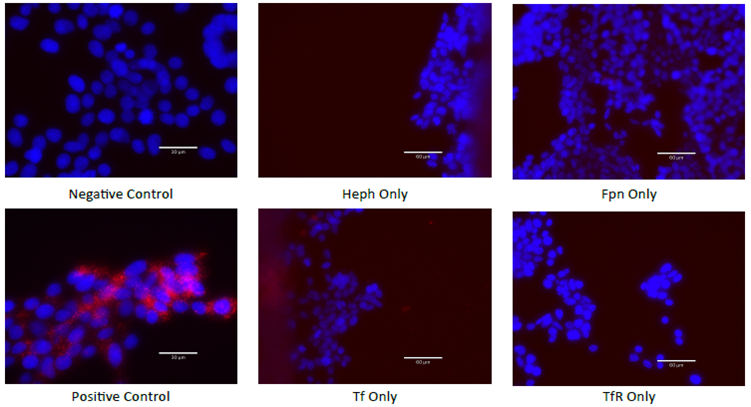
To complete the process of iron export, Fpn works in a complex with many proteins, including Heph10,11. Heph is a ferroxidase that converts the Fpn-exported ferrous (Fe2+) to ferric (Fe3+) that can bind to apo-Tf and be utilized by cells. Numerous studies have shown that Heph is required to stabilize Fpn in the plasma membrane and to enable iron export10,11,28,29. We have replicated these findings, by demonstrating that Fpn and Heph can be co-immunoprecipitated from ECs. Furthermore, we demonstrate the novel finding that both apo- and holo-Tf independently are co-immunoprecipitated with Fpn and Heph. These results suggest that apo- and holo-Tf bind to Fpn and Heph in a complex of iron export proteins. In order to narrow down which protein holo-Tf bound to in the membrane that resulted in decreasing Fpn, we employed PLA. We found that holo-Tf directly interacts with Fpn, while apo-Tf does not. On the other hand, apo-Tf interacts with Heph, while holo-Tf does not, a finding that is supported in the literature16,18,30. It is hypothesized that apo-Tf binds to Heph to accept the ferric iron that Heph converts from ferrous iron. This stimulates the release of more iron as long as there is apo-Tf to accept it. Taken together these data suggest that apo- and holo-Tf differentially interact with iron export proteins, likely due to their structural differences31. The exact binding sites, conformation changes, and catalysts for these interactions are an exciting unexplored area that could pave the way for clinical manipulation. For example, as has been done experimental7, Tf could be infused to modulate iron accumulation in diseases in which it is dysregulated. Additionally, pharmaceuticals could be designed to facilitate or inhibit the endogenous protein interactions in an effort to correct brain iron accumulation.
Prior to the discovery that elevated holo-Tf could suppress iron release, hepcidin was the primary focus of iron release regulation13. Hepcidin is a pro inflammatory hormone peptide primarily secreted by the liver and upregulated in environments of inflammation and high iron levels32. Astrocytes33,34 and the choroid plexus35,36 have also been shown to secrete hepcidin, though in much smaller amounts that cannot account for total brain hepcidin levels36,37, suggesting much of the brain hepcidin comes from systemic levels when pathologically necessary, though this has not yet been proven. A number of groups have shown that astrocytic hepcidin reduces Fpn levels and subsequent iron release14,38,39. However, we have previously demonstrated that supraphysiological levels of hepcidin are not capable of blocking iron release from ECs3,4. These data suggest that hepcidin cannot be the sole regulator of iron release in the brain. In support of this notion, Enculescu et al. modeled iron levels, and when compared to their experimental results, the study found that hepcidin control over iron uptake was necessary, but not sufficient40. Once a secondary regulatory mechanism was added to the model, their experimental results aligned with the model40. Thus, our data directly support that hepcidin is not the sole regulator of iron release and indicate the additional regulators are apo- and holo-Tf.
Our data offer an opportunity to explore the concept of regulation of iron uptake in general by hepcidin. We found that hepcidin competes with holo-Tf for binding to Fpn at low holo-Tf and high hepcidin concentrations. However, when there was more holo-Tf or less hepcidin present, this effect was reduced. Notably, when hepcidin was only present at physiological baseline levels26, there was no interruption of the interaction between holo-Tf and Fpn. These findings suggest that hepcidin is only effective at controlling Fpn levels at levels consistent with inflammation or high iron. In observing competition between holo-Tf and hepcidin for Fpn binding, the internalization of Fpn was inhibited to determine if the competition was for binding site availability or rate of internalization. By preventing the internalization of Fpn, hepcidin had no impact on the interaction between holo-Tf and Fpn. This suggests that hepcidin internalizes Fpn faster than holo-Tf, which was confirmed by isolating membrane Fpn. Hepcidin reduces membrane Fpn by nearly 50% in 5 minutes, whereas holo-Tf only starts to reduce membrane Fpn at 60 minutes. On the other hand, no amount of hepcidin impacts the interaction between apo-Tf and Heph. These data offer the intriguing suggestion that if apo-Tf is present, it will bind to Heph even in pathological states and may be an explanation for iron accumulation in neurodegenerative disease. It has been postulated that in Alzheimer’s disease41 and Parkinson’s disease42 the brain may start as functional iron deficient, along with elevated levels of apo-Tf, which triggers increased iron uptake until the excess iron detrimentally damages the BBB and surrounding cells. The question remains however, if the binding of apo-Tf to Heph will continue to stimulate iron release in the presence of hepcidin.
The model of apo- and holo-Tf regulation of iron release from ECs works as a feedback loop. As cells, such as neurons or astrocytes, need iron for metabolic processes, myelin synthesis, or dopamine synthesis, they take up holo-Tf through TfR43. Once endocytosed, the iron is removed and the resulting apo-Tf is released43. The communication of brain iron status via apo- and holo-Tf allows cells to signal their iron needs based on their iron consumption. Numerous studies have shown higher regional iron uptake that correspond to areas with higher iron needs9,44,45. Our pervious data suggest that as the apo- to holo-Tf ratio changes in the extracellular fluid, more iron is released locally from the BBB. In support of this notion are data showing CSF from iron deficient monkeys and iron chelated astrocytes increase iron release from cultured bovine retinal ECs (BRECs), while iron loaded biological samples resulted in decreased iron release6. These data have been replicated when cells are exposed to apo- or holo-Tf directly3,4,6 or when apo- or holo-Tf is directly infused into the brain7. In all studies mentioned here, apo-Tf increased iron release while holo-Tf decreased iron release.
The data in this study expand the model for brain iron uptake by suggesting that apo-Tf stimulates iron release by binding to Heph to access exported free iron (Fig. 6A). Once loaded with iron, the now holo-Tf becomes available to surrounding cells. If the levels of holo-Tf in the extracellular fluid rise, holo-Tf binds to Fpn to suppress more iron release (Fig. 6B). The internalization of Fpn by holo-Tf is not rapid, unlike hepcidin. When upregulated and present in high amounts, hepcidin can rapidly internalize Fpn (Fig. 6C). Thus, we propose that hepcidin is likely used as a fast acting, immediate stop to iron release in environments of inflammation and very high iron. However, for moment-by-moment regional control of iron release, holo-Tf may be a better candidate to regulate regional iron supply
{kind=link}
{kind=link}
{kind=link}