Exposure to VLH activated the gene expression in leukocytes
Microarray data of GSE103297 was based on 112 PBMC samples collected from 21 subjects at seven time points of VLH exposure. At baseline, subjects were studied at sea level (130 m, Pb = 749 mmHg). At d1noon or d1pm, d7, d16, post7, or post21, subjects were ascended or reascended to the target altitude (5260 m, Pb = 406 mmHg), sampled, and studied (Fig. 1A). Laboratory values concerning PaO2 (mmHg), PaCO2 (mmHg), SaO2 (%), CaO2 (ml/dL), P50 (mmHg), hemoglobin (g/dL), LLQ-AMS score, and AMS-C-Composite score were detected and recorded. PCA was conducted to determine the timeline differences in the gene expressions (Fig. 1B). Gene expression patternes at d1pm and d7 indicated an apparent distinction from baseline (increasing along the PC1 axis), but this trend reduced both along the PC1 and PC2 axes at d16 and post7, suggesting the acute exposure to VLH may change the gene expression pattern in monocytes. Then the DGs across the baseline, d1pm, and d7 were determined by P value (< 0.05) and Log2FC (> 1.3 or < -1.3). There were 512 overlapped genes with consistent significance along with the exposure timeline from d1pm to d7 (Fig. 1C, Table S2). We then conducted GO analysis to identify the function of the 512 DGs, and it was indicated that most of them were involved in leukocyte activation (Fig. 1D). 12 DGs enriched in the GO term “leukocyte activation involved in immune response” were further explored for the expression level along the timeline (Fig. 1E). It was indicated that these leukocyte activation-related genes were up-regulated along the timeline with a peak at d7, suggesting leukocyte was activated upon VLH, but it would recover along the exposure timeline.
T Cells Dominated The Genetic Responses Upon Vlh Exposure
To investigate the functional differenation of leukocytes upon VLH exposure, 1644 genes (Table S3) with significant difference from the baseline were functionally clustered along the timeline, and six clusters were yielded (Fig. 2A). Both cluster 1 (299 genes) and cluster 6 (384 genes) were inversely changed across the timeline, with peaked decrease in cluster 1 while increase in cluster 6 at d7. Different expression dynamics were also observed in cluster 2 (213 genes), cluster 3 (281 genes), cluster 4 (123 genes), and cluster 5 (344 genes). The function of each cluster was annotated using LM22[22] (Fig. 2B). As expected, each cluster was functionally associated with different leukocyte types, with T cells CD4/CD8 dominating in cluster 1/2/4/5, T cells regulatory in cluster 4/5, T cells gamma delta in cluster 2/4/5/6, NK cells resting in cluster 6, monocytes in cluster 2/6, dendritic cells activated in cluster 3, and neutrophils in cluster 6. We then compared the biological functions among the clusters using GO, KEGG, Reactome, and WikiPathway database (Fig. 2C). No significant terms were enriched in cluster 5. No relationship was observed in the expression pattern between cluster 2, 3, and 4. Both cluster 1 and 6 overlapped in T cell-associated functions, oxidative stress, and blood coagulation. Cluster 1 was specifically enriched in cell death in response to hydrogen peroxide and regulation of response to reactive oxygen species. Cluster 6 was specifically enriched in platelet activation. Both cluster 1 and 6 were indicated with dramatic altitude or hypoxia sensitivity, and both were functionally associated with T cell activities, suggesting the dominant roles of T cells in the genetic responses to VLH exposure.
Inverted Cd4/cd8 Ratio May Function As The Risk Factor Of Sams
To investigate the timeline abundence of various leukocytes in subjects exposed to VLH, cibersort algorithm[23] was applied in combination with LM22, and the expression profiling at baseline, d1pm, d7, d16, and post7 (Fig. 3A and Table S4). T cells CD8, CD4, NK cells resting, and monocytes were indicated as the major cell types in PBMC samples, with significant decrease in the estimated proportion of CD4 cells at d1pm and d7 vs. baseline, which resulted in the inverted CD4/CD8 ratio at d1pm and d7 (Fig. 3B, Table S5). When predicted using ROC, SaO2 (AUC = 0.833), P50 (AUC = 0.833), and the serum level of hemogbin (AUC = 0.792) performed well to differeniate subjects with normal or inverted CD4/CD8 ratio (Fig. 3C), suggesting their potential effects in CD4/CD8 banlance. SaO2 (AUC = 0.617), CaO2 (AUC = 0.783), P50 (AUC = 0.667), hemogbin (AUC = 0.850), and the estimated proportion of T cells (AUC = 0.633) were also indicated as the possible binary classifiers for sAMS (sAMS or non-sAMS) (Fig. 3D). The interplay effects between CD4/CD8 ratio, SaO2, P50, hemogbin, the estimated proportion of T cells, and CaO2 implied that, the inverted CD4/CD8 ratio may function as the potential risk of sAMS.
Genetic Profiling For Sams
To uncover the gene signature underlying the phenotypes of sAMS, subjects in the training cohort were binarily subgrouped with the best predicted thresholds of various classifiers. The interplay effects between classifiers were investigated regarding the expression pattern of DGs across the binary subgroups (Fig. 4A). Significant correlations were observed between SaO2, P50, hemoglobin, and CD4/CD8 ratio in the expression pattern of related DGs (P < 0.001), meanwhile, as the potential risk factors of sAMS, CD4/CD8 ratio, the estimated proportion of T cells, SaO2, P50, hemoglobin, and CaO2 were indicated with significant correlations with LLQ-AMS score (P < 0.001). 2291 risk factor-related DGs were identified, with 328 in the set of CaO2, 346 in CD4/CD8 ratio, 682 in hemoglobin, 964 in P50, 515 in SaO2, 263 in the estimated proportion of T cells, and 508 in LLQ-AMS score (Table S6), which were intersected in 2 to 6 ways under the Venn analysis (Fig. 4B). To further identify function modules among the 2291 DGs and the relationship to the risk factors of sAMS, WGCNA was performed using the values of PaO2, PaCO2, SaO2, CaO2, P50, hemoglobin, LLQ-AMS score, AMS-C-composite score, CD4/CD8 ratio, and the estimated proportion of T cells as trait. Gene co-expression network was constructed and 7 modules were identified using the tools of hierarchical clustering, dynamic tree cut, and merged dynamic (Fig. 4C). Next, we established the module-trait relationships (Fig. 4D). The royalblue module was negatively correlated with SaO2, but positively with CD4/CD8 ratio. Both purple and pink modules were negatively correlated with hemoglobin, while positively with LLQ-AMS score and AMS-C-composite score. Morever, the purple module seemed to be sensitive to CaO2, likewise, blue to hemoglobin, turquoise to P50, tan, yellow and turquoise to CD4/CD8 ratio and red, cyan to LLQ-AMS score. The results demonstrated that SaO2, CaO2, P50, hemoglobin, LLQ-AMS score, AMS-C-composite score, CD4/CD8 ratio, and the estimated proportion of T cells, as the potential risk factors of sAMS, may impact the disease outcome at the genetic level.
Deep Learning To Establish The Prediction Model Of Sams
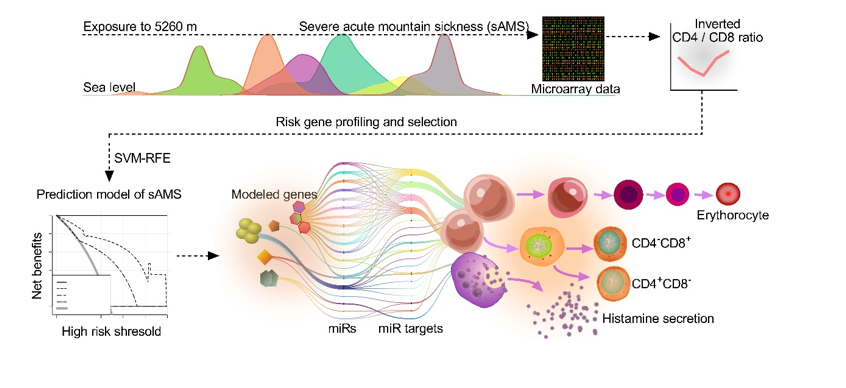
To identify the marker genes of sAMS, we contructed a prediction model of sAMS using the deep learning of SVM-RFE, which consists of the classification algorithm and the feature selection algorithm wrapped around, strategized to select or remove some features from the high-dimensional feature set, and obtain the optimum feature subset from various candiate subsets generated. Therefore, SVM-RFE is actually designed to find a hyperplane of the maximized marginal distance with the best differentiating performance between the two categories of the dataset, which is represented with the weight vector WT, feature vector X, and the threshould b as following: WTX + b= 0 (Fig. 5A). Obviously, when WTX + b= 0, the sum of the marginal distances from the hyperplane to the closest features (D1 + D2) is maximized, however, it would be indicated with the poorest differentiating performance and accuracy whenever WTX + b= 1 or -1. In deep learning of SVM-RFE (Fig. 5B), the initial features (M = 2291) were input for classifier-training, with the relevance of the n–th entry of X determined by the corresponding value Wn in WT(n = 1, 2,…M). Then, in each fold (k = 15) of CV, the concrete number of features (τ = 30) with the lowest absolute values of Wn were rejected. The max accuracy was determined by the entire feature selection and error estimation process (five-fold CV). The top 14 ranked features (Table S7) with the highest five-fold CV accuracy (Fig. 5C) or the lowest error (Fig. 5D) were selected for further analysis. 10 of 14 with significant predictive power (P < 0.05) (Table 1) were used to build the model (C-index = 1, P < 0.01) (Table S8) and nomogram (Fig. 5E). In the training cohort of d1, the model was indicated with excellent prediction performance for sAMS as analysed using ROC (AUC = 1) (Fig.S1A), calibration curve (Fig. 5F), and survival analysis (R2 = 5.011, P = 0.024) (Fig. 5G). When tested using AUC within the timeline of the training cohort over baseline (AUC = 0.600), d7 (AUC = 0.691), d16 (AUC = 0.673), post7 (AUC = 0.633), and validated in the validation cohort (AUC = 0.626) (Fig. S1B-F), the model was indicated with satisfactory predictive accuracy between the actual probability and the predicted. To assess the clinical applicablity of the model, we also established a single-gene (OR10G8) model (C-index = 0.764) using the baseline data of the training cohort (Fig S2, Table S9), and a three-gene model (B4GALT4, DIP2B, GALNT13) (C-index = 0.897) (Fig S3) based on the validation-cohort data (Table S10, S11). All the models were indicated with overall net benefits varing from 53–100% when assessed using DCA (Fig. 5H).
Table 1
Univariate logistic regression analysis
| Dependent variables | Coefficient | Standard error | Statistics | OR | | P value |
| ACSM1 | 2.639 | 1.126 | 2.340 | 14.001 | | 0.019 |
| B4GALT4 | 4.277 | 1.495 | 2.860 | 72.002 | | 0.004 |
| DHX58 | 2.639 | 1.126 | 2.340 | 14.001 | | 0.019 |
| DIP2B | 2.639 | 1.126 | 2.340 | 14.001 | | 0.019 |
| EPHB3 | -3.466 | 1.323 | -2.620 | 0.031 | | 0.009 |
| GALNT13 | 2.639 | 1.126 | 2.340 | 14.001 | | 0.019 |
| IFNA5 | -1.099 | 0.957 | -1.150 | 0.333 | | 0.251 |
| MARVELD3 | 4.277 | 1.495 | 2.860 | 72.002 | | 0.004 |
| OR10G8 | -2.100 | 1.058 | -1.990 | 0.122 | | 0.047 |
| OR5B3 | 4.277 | 1.495 | 2.860 | 72.002 | | 0.004 |
| RHEBL1 | -3.466 | 1.323 | -2.620 | 0.031 | | 0.009 |
| SLC8A2 | 1.540 | 0.988 | 1.560 | 4.666 | | 0.119 |
Micrornas (Mirs) Mediated The Effects Of The Featured Genes In The Development Of Sams
To further explore the roles of the 14 featured genes in the development of sAMS, miRs were predicted using the miR function of FunRich (version 3.1.3). There are 29 homo sapiens (hsa)-miRs identified in five featured genes, including 2 from EPHB3, 3 from DIP2B, 8 from RHEBL1, 3 from GALNT13, and 13 from SLC8A2, which were targeted to 3710 miR targets (Fig. 6, Table S12). We next wanted to know the biological functions of the miR targets. As expected, most targets were enriched in terms related to lymphocyte activities under GO analysis (73.69%) (Fig. 7). Furthermore, there were 5.26% enriched in histamine secretion, 2.56% in erythrocyte differentiation, and 15.79% associated with the regulation of myeloid cell differentiation (Table S13). Accordingly, several meaningful pathways of the featured genes were identified, including GALNT13-(hsa-miR-124-3p/ 506-3p)–RCOR1 (Fig. 8A), SLC8A2/ DIP2B-(hsa-miR-133a-3p/ 133b)-RVAMP2/ SLC4A1 (Fig. 8B and C), RHEBL1-( hsa-miR-19a/ b-3p)-HIF1A, and EPHB3-(hsa-miR-149-5p)-IL6, which were functionally related to erythrocyte differentiation, alpha-beta T cell differentiation, and histamine secretion by mast cells (Fig. 9). These results suggested the important roles of the featured genes on sAMS, which were mediated by miRs and their downstream targets.
{kind=link}