Cell Culture
The 4T1 cells (CRL-2539, mouse triple-negative mammary tumor cell line isolated from Balb/c mice), MDA-MB-231 cells (HTB-26™, human triple-negative mammary tumor cell line) and RAW 264.7 cells (TIB-71, macrophage-like cell line derived from Balb/c mice) were obtained from American Type Culture Collection (ATCC). 4T1 cells and RAW 264.7 cells were cultured in Roswell Park Memorial Institute Medium (RPMI) and MDA-MB-231 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and a penicillin/streptomycin cocktail (100 µg/mL).
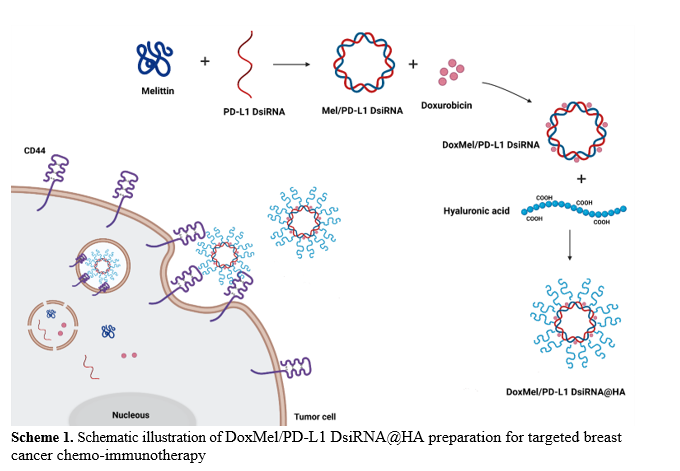
Construction Of Mel/pd-l1 Dsirna
Briefly, 100 µl DsiRNA (Integrated DNA Technologies) at a final concentration of 10 µM was added to 100 µl Mel (> 85% in purity, M2272, Sigma-Aldrich) at different Mel/DsiRNA ratios (0.5:1, 1:1, 2:1, 3:1 and 4:1). The mixture was then incubated for 1 h in room temperature, followed by electrophoresis using 2.5% agarose gel to assess the formation of Mel/PD-L1 DsiRNA.
Stability Of Mel/pd-l1 Dsirna
The stability of Mel/PD-L1 DsiRNA was evaluated in the presence of RNase. In brief, RNase (R1253, ThermoFisher) was added into solutions containing either PD-L1 DsiRNA or Mel/PD-L1 DsiRNA. After 2-hour incubation at room temperature, the structure of PD-L1 DsiRNA and Mel/PD-L1 DsiRNA was evaluated by applying 2.5% agarose gel electrophoresis.
Preparation Of Doxmel/pd-l1 Dsirna
Twenty microlitre of Dox (15007, Cayman Chemical) at a final concentration of 20 µM was added to the solution with increasing concentrations of Mel/PD-L1 DsiRNA. Following incubation at room temperature for 2 hours, the amount of Dox loaded onto Mel/PD-L1 DsiRNA was determined through measuring the fluorescence of Dox at the excitation of 480 nm using SpectraMax iD3 microplate reader.
Preparation Of Of Doxmel/pd-l1 Dsirna@ha
To generate DoxMel/PD-L1 DsiRNA@HA, 300 µl HA (ab143634, Abcam) at a concentration of 10 mg/ml was added to 1 ml of DoxMel/PD-L1 DsiRNA solution and stirred for 3 hours. After that the final product (DoxMel/PD-L1 DsiRNA@HA) was collected by centrifugal device. The size and zeta potential of collected particle and other intermediate products were assessed by dynamic light scattering (DLS, Malvern Zetasizer). Moreover, transmission electron microscopy (TEM, HT-7700, Hitachi, Japan) was applied to observe the morphology of DoxMel/PD-L1 DsiRNA@HA.
Drug Release Study
The effect of pH on Dox release was measured using centrifugal devices. Briefly, DoxMel/PD-L1 DsiRNA@HA was immersed into phosphate buffer saline and sodium acetate buffer (pH 7.4 or 5.5, respectively). At specific time points (1, 3, 6, 12, 24 and 36 hours), DoxMel/PD-L1 DsiRNA@HA was collected from the buffer by 3K centrifugal device and the fluorescent intensity of buffer, which represents the amount of the released Dox, was measured using SpectraMax iD3 microplate reader. The accumulative release percentage (AR %) of Dox was calculated by applying the equation of AR % = D1/D0 × 100, where D1 is the concentration of released Dox and D0 is the concentration of Dox loaded onto DoxMel/PD-L1 DsiRNA@HA.
Protein Expression Of Cd44 In Tnbc Cells
To measure the expression level of CD44, RAW264.7, 4T1 and MDA-MB-231 cells were subjected to western blot analysis using CD44 antibody (15675-1-AP, Proteintech).
Measurement Of Pd-l1 Expression
The 4T1 cells were cultured in 24-well plates and treated with Mel/PD-L1 DsiRNA and Mel/PD-L1 DsiRNA@HA (final concentration of siRNA for both treatments was 200 nM). After incubation for 48 hours, cells were lysed in buffer (10 mm HEPES pH 7.4, 50 mm Na pyrophosphate, 50 mm NaF, 50 mm NaCl, 5 mm EDTA, 5 mm EGTA, 100 µm Na3VO4, and 0.1% Triton X-100) and the expression of PD-L1 was evaluated by western blotting using PD-L1 antibody (66248-1-Ig- Proteintech).
In vitro cytotoxicity assay
The 4T1 cells were seeded onto 96-well plates with 5 × 103 cells per well (n = 5). The next day, cells were treated with Dox, Mel, DoxMel/PD-L1 DsiRNA and DoxMel/PD-L1 DsiRNA@HA. The concentrations of Dox and Mel were 1 and 2 µM in each treatment, respectively. After 24 and 48 hours of treatment, cells were incubated with 10 µl of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) solution (G9243, Promega) for 3 hours. Finally, a microplate reader (BioTek Synergy H1) was employed to measure absorbance of each sample at 490 nm. The optical density (OD) values of sham cells were set at 100% viability, and the percentage of viability of treated cells was calculated accordingly.
Apoptosis Analysis
To evaluate the impact of the developed treatment on apoptosis, 4T1 cells were seeded onto 12-well plates for 24 hours, followed by incubation with Dox, Mel, DoxMel/PD-L1 DsiRNA and DoxMel/PD-L1 DsiRNA@HA. The concentrations of Dox and Mel were 1 and 2 µM in each treatment, respectively. After 24 hours, cells were collected and resuspended in annexin binding buffer (V13246, Thermofisher Scientific). Subsequently, 5 µl of the annexin V- Fluorescein isothiocyanate (FITC) staining (A13199, Thermofisher Scientific) reagent was added to the cells, followed by incubation in dark for 20 minutes. Finally, flow cytometry was employed to analyze the stained cells. Results were analyzed with FlowJo version 10 software.
Induction Of Immunogenic Cell Death (Icd)
The exposure of cellular calreticulin was examined to assess the effect of the designed treatment on ICD. Briefly, 4T1 cells were seeded onto a 12-well plate and treated with Dox, Mel, DoxMel/PD-L1 DsiRNA and DoxMel/PD-L1 DsiRNA@HA. The concentrations of Dox and Mel were 1 and 2 µM in each treatment, respectively. After 24 hours, cells were collected and incubated with calreticulin (Alexa Fluor® 647) antibodies (ab196159, Abcam) at 4°C in the dark for 1 hour. Then cells were washed and subjected to flow cytometry analysis.
The amount of extracellular high mobility group box 1 (HMGB1) protein after each treatment was evaluated by western blotting. In brief, supernatant of cells after treatment was collected. An equal volume of methanol and 0.25 volume of chloroform was added to each supernatant. After mixing them thoroughly, samples were centrifuged for 10 mins at 20,000 ⋅ g. The upper phase was discarded and 500 µl methanol was added into the interphase, followed by another round of centrifugation for 10 mins at 20,000 × g. At the end, protein pellet was dried at 55oC, resuspended in protein loading buffer and western blotting was performed for evaluation of HMGB1.
Furthermore, RealTime-Glo™ extracellular ATP assay kit (GA5010, Promega) was applied to measure the level of released ATP into the supernatant. For this aim, after incubation of cells with each treatment for 36 hours, 20 µl of 4× RealTime-Glo™ extracellular ATP assay reagent was added into each well, and luminescence was measured employing a microplate reader (BioTek Synergy H1).
Animal Studies
Female Balb/c mice (aged 6–8 weeks, 000651, The Jackson Laboratory) were used for animal studies. All animal experiments described below were performed under the guidance and approval of Animal Care Committee at the University of British Columbia (A18-0275).
In vivo anti-tumor performance
The 4T1 cells (5 × 105 cells) were subcutaneously implanted into the right flank of mice to establish tumors. After tumors reached ~ 50 mm3, mice were randomly separated into 5 groups: 1- mice received PBS as control, 2- mice received Dox, 3- mice exposure to Mel, 4- mice received DoxMel/PD-L1 DsiRNA, 5- mice treated with DoxMel/PD-L1 DsiRNA@HA. All mice received intratumoral injection twice on day 0 and 4. The concentration of Dox, Mel and PD-L1 DsiRNA was the same for all groups and detailed as follows: 2 mg/kg Dox, 4 mg/kg Mel, and 1 mg/kg PD-L1 DsiRNA. Following treatment, tumor size was measured using a digital caliper and calculatedthrough the following equation: (volume = length × width2 × 0.52). Tumor suppression rate (TSR) was measured as TSR (%) = [1 − (tumor volume of the treated group)/(tumor volume of the control group)] × 100 (%). Mice were euthanized once they showed any of the following signs according to our protocol: ≥ 20% of their initial body weight loss, ulceration observed in ≥ 10% of tumor region, ≥ 1.7 cm of tumor size in diameter or ≥ 10% of tumor weight over body weight. Furthermore, to investigate the efficacy of each treatment, the survival rate of each group was documented. At the end of the experiment, tumor tissues were collected to examine the rate of apoptosis using Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay based on the manufacturer’s protocol (G3250, Promega).
Safety Evaluation Of In Vivo Treatments
To evaluate the safety profile of each designed treatment, body weight of mice was recorded every three days until the end of the experiment. Moreover, at 15 days post treatment, a different group of mice (n = 4) was euthanized and main tissues were collected for the assessment of potential drug toxicities by hematoxylin-eosin (H&E) staining.
Analysis Of Pd-l1 Expression And Immune Response
The expression of PD-L1 in tumor tissues was assessed through immunofluorescence staining. In brief, mouse 4T1 tumors were collected at 15 days post-treatment, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked with 10% FBS. Tumors were then subjected to staining with PD-L1 antibody overnight, followed by incubation with Alexa Fluor® 488-conjugated secondary antibody (A11029, Invitrogen) for 1 hour. Finally, tumors were mounted with DAPI and the slides were scanned under Zeiss LSM 880 inverted confocal microscopy. For analysis of infiltration of immune cells into tumor microenvironment, tumor sections were deparaffinized, rehydrated, and then stained with anti-CD8 (sc-1177, Santa Cruz Biotechnology), anti-NK1.1 (14-5941-82C, eBioscience), and anti-F4/80 (sc-377009, Santa Cruz Biotechnology) antibodies. Immunohistochemistry (IHC) was performed using the MACH4 Universal HRP-Polymer Detection System (BRI4012H, Biocare Medical) and hematoxylin solution Gill II (GHS232, Sigma-Aldrich) as previously noted [24], Lastly, Aperio ScanScope AT (Digital slide scanner, Leica Biosystems Inc) was employed to obtain whole-slide digital images. Quantification analysis was conducted using NIH ImageJ (version 1.52p) and presented as relative optical density. In addition, the level of proinflamatory cytokines, including IFN-γ, IL-6 and TNF-α, as well as granzyme B in tumor microenvironment was evaluated via immunofluorescence staining using anti-IFN-γ (505802, Biolegend), anti-IL-6 (504502, Biolegend), anti-TNF-α (506302, Biolegend), and granzyme B antibody (4275, Cell signaling).
Distant Tumor Study
A bilateral tumor model was established to assess the impact on distant tumors. Briefly, 4T1 cells (2 × 105 cells) were subcutaneously injected into the right flank of mice. Three days later, mice were rechallenged with another subcutaneous injection of 4T1 cells (2 × 105 cells) into the left flank. Once primary tumors on the right flank reached ~ 80mm3, mice were received intratumoral (right flank) injection of different formulations (as above). The length and width of the distant tumors were monitored every three days using a digital caliper.
{kind=link}