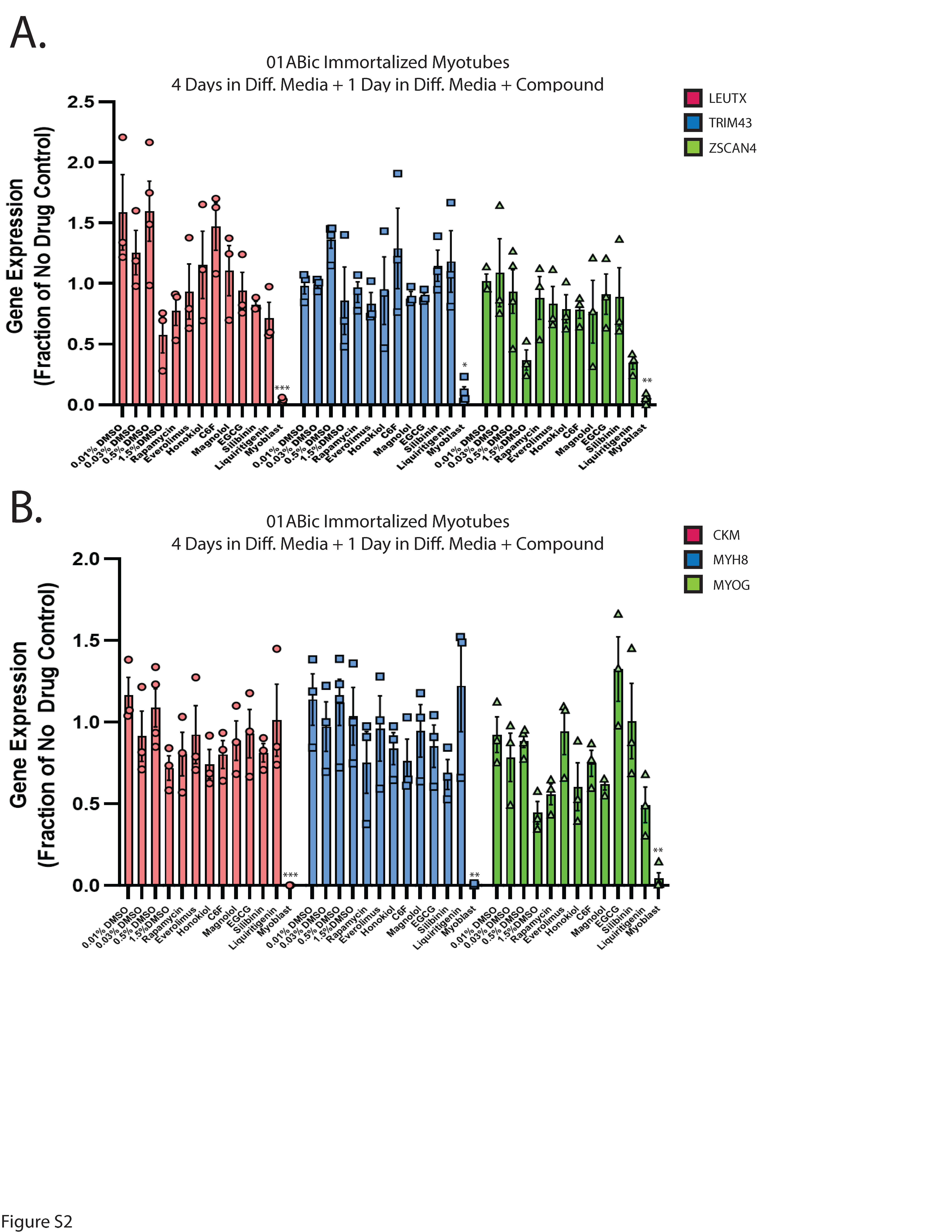
First-generation small molecules inhibit DUX4-induced apoptosis. In our previous work, we demonstrated that the hyaluronic acid synthesis inhibitor 4MU can provide resistance to DUX4-induced toxicity (36). Additionally, we and others have implicated hypoxia signaling as central to toxicity (41, 52). Interestingly, these signaling pathways converge on the mTor/AKT/PI3K signal transduction axis (71, 72), and inhibition of this pathway can provide resistance to toxicity (41). We sought to leverage these observations to identify small molecules that can provide resistance to DUX4-induced apoptosis. Unfortunately, 4MU itself requires millimolar doses for maximal effectiveness, and so is not suitable for use as a therapeutic. Thus, we considered other molecules that may provide a similar result at lower concentrations. Based on previous reports of their activity in a relevant pathway, we identified a panel of six first-generation compounds that had the potential to meet this criteria- honokiol (Cas # 35354-74-6), its synthetic analogue C6F, magnolol (Cas # 528-43-8), epigallocatechin gallate (EGCG, Cas # 989-51-5), silibinin (CAS # 22888-70-6), and liquiritigenin (Cas # 69097-97-8) (73–77). Notably, EGCG, silibinin, and liquiritigenin were of particular interest because their chemical structures are built around the same fused ring structure as 4MU, and they each maintain the chemically active hydroxyl group (78) (Fig. 1A). To evaluate these compounds, we used the MB135-DUX4i myoblast model (38). Myoblasts were seeded on 96 well plates, and the following day they were pre-treated by adding the indicated compound to the media for three hours, followed by the addition of 2 mg/mL doxycycline (DOX) to the media for 24 hours to induce DUX4 expression (for a total of 27 hours of exposure to the compounds). As a positive control, we also included the mTor inhibitor rapamycin, which can inhibit DUX4-induced toxicity (41) and its next-generation analogue everolimus. Cell death was then visualized using the CellEvent Caspase 3/7 Green assay (Invitrogen, Waltham, MA USA). We tested a range of concentrations and found that each was able to provide resistance to toxicity when administered at proper concentrations (Fig. 1B, Figure S1). To confirm and quantitate these results, we conducted similar experiments using the Caspase-Glo 3/7 assay system (Promega, Madison, WI USA). Each compound provided at least a twofold reduction in caspase 3/7 activity relative to vehicle controls, with liquiritigenin showing the strongest effect (Fig. 1C). To validate these results we performed limited-cycle RT-PCR as described previously (41) and confirmed that these compounds did not interfere with the induction of the codon-altered DUX4 transgene (Fig. 1D). Similarly, we performed western blotting analysis to determine the effects on the levels of DUX4 protein. As observed previously (41), rapamycin caused a drop in the abundance of DUX4 (Fig. 1E). Surprisingly however, only honokiol showed a similar decline in DUX4 protein abundance, but the remaining compounds had no effect. This confirms that the mechanism of observed resistance to DUX4-induced toxicity occurs downstream of DUX4 expression, but also unexpectedly suggests that these compounds function via a different mechanism than rapamycin. Finally, to confirm that these compounds function downstream of DUX4 expression and that they do not have a deleterious effect on mature myotubes, immortalized patient-derived 16ABic myoblasts (79) were induced to form myotubes for 4 days using established methods (80), and were then treated with compounds for 24 additional hours. The expression of three DUX4-target genes were then analyzed using qRT-PCR, and no significant change was observed (Fig. 2A). We also analyzed three myogenesis markers and found no statistically significant effect on their expression (Fig. 2B). Similar results were observed using a second patient-derived cell line (Figure S2).
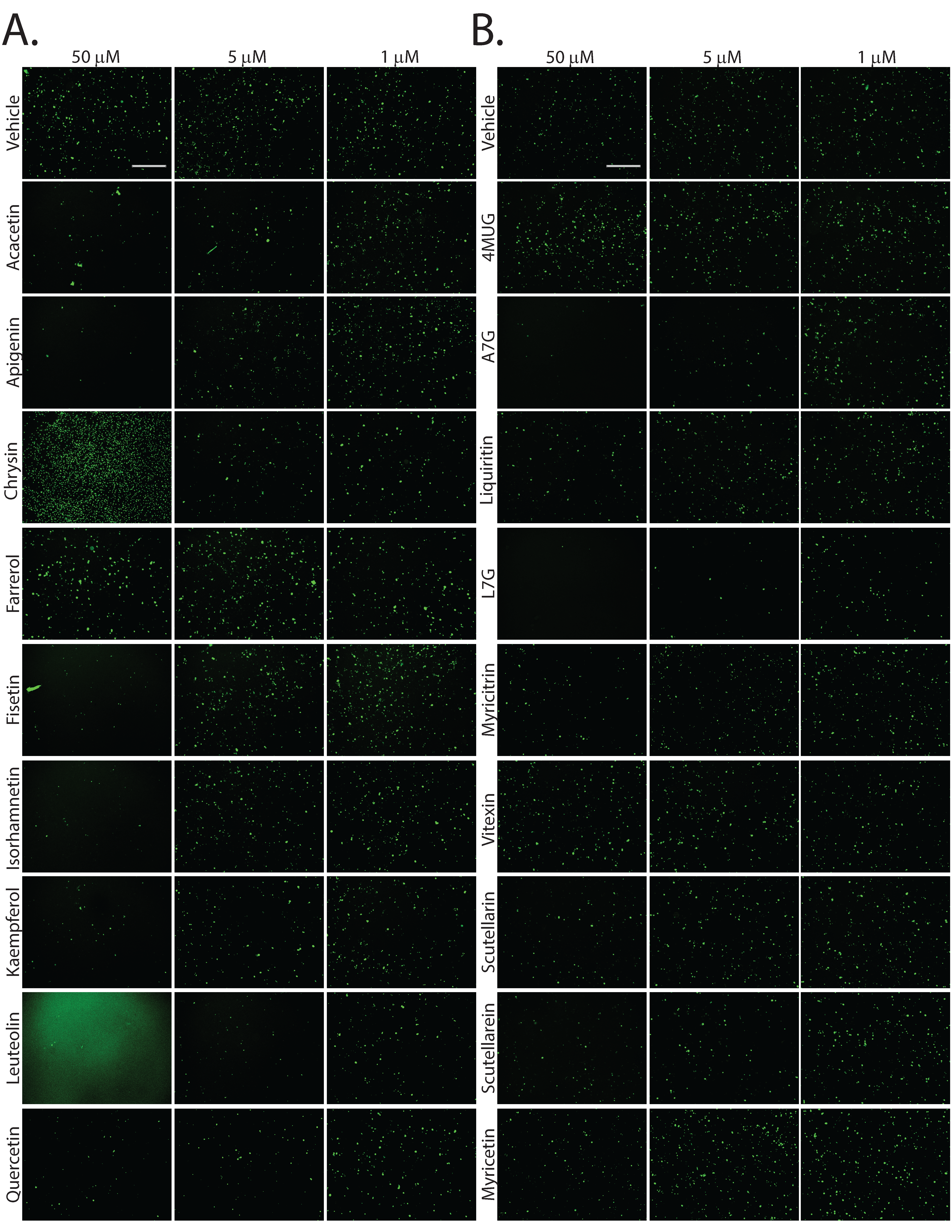
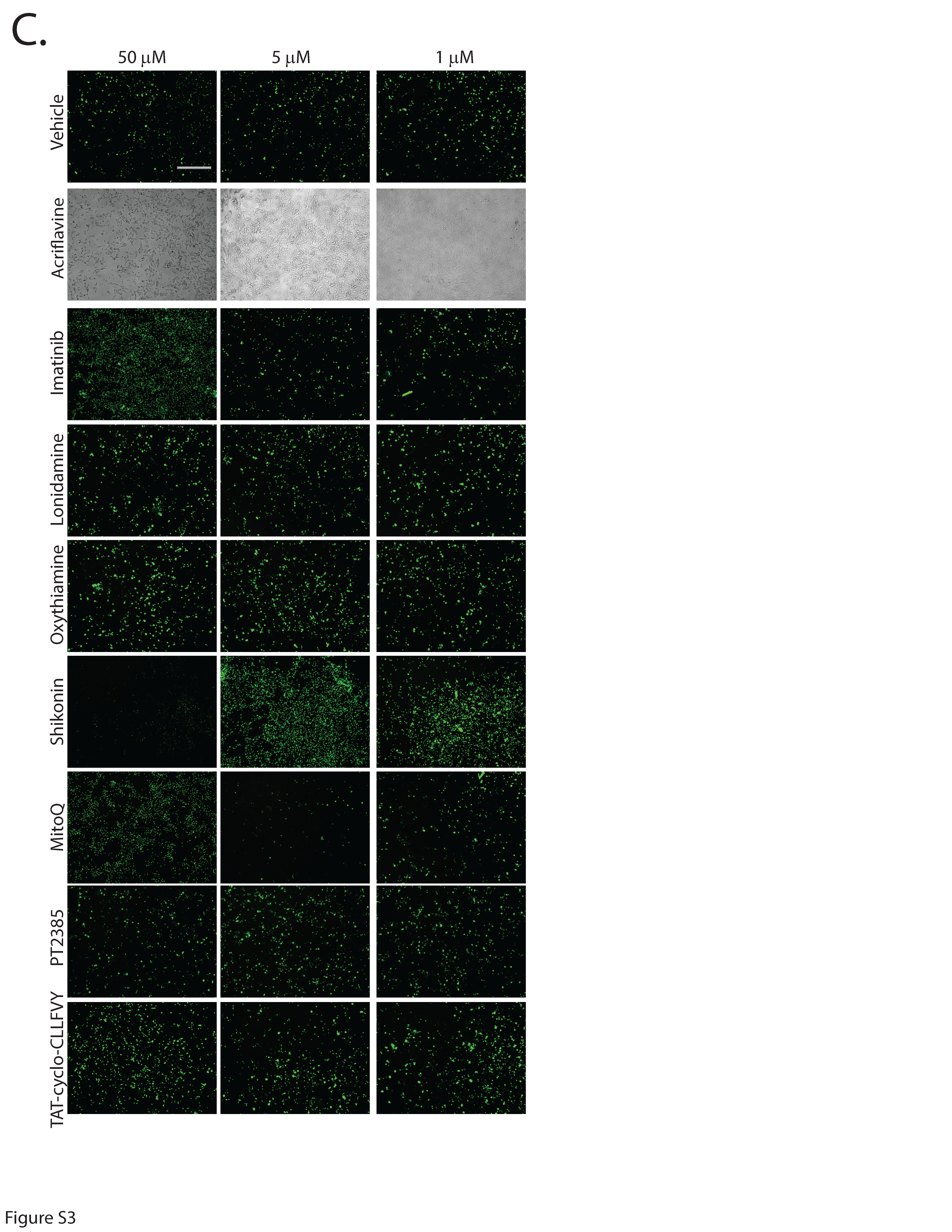
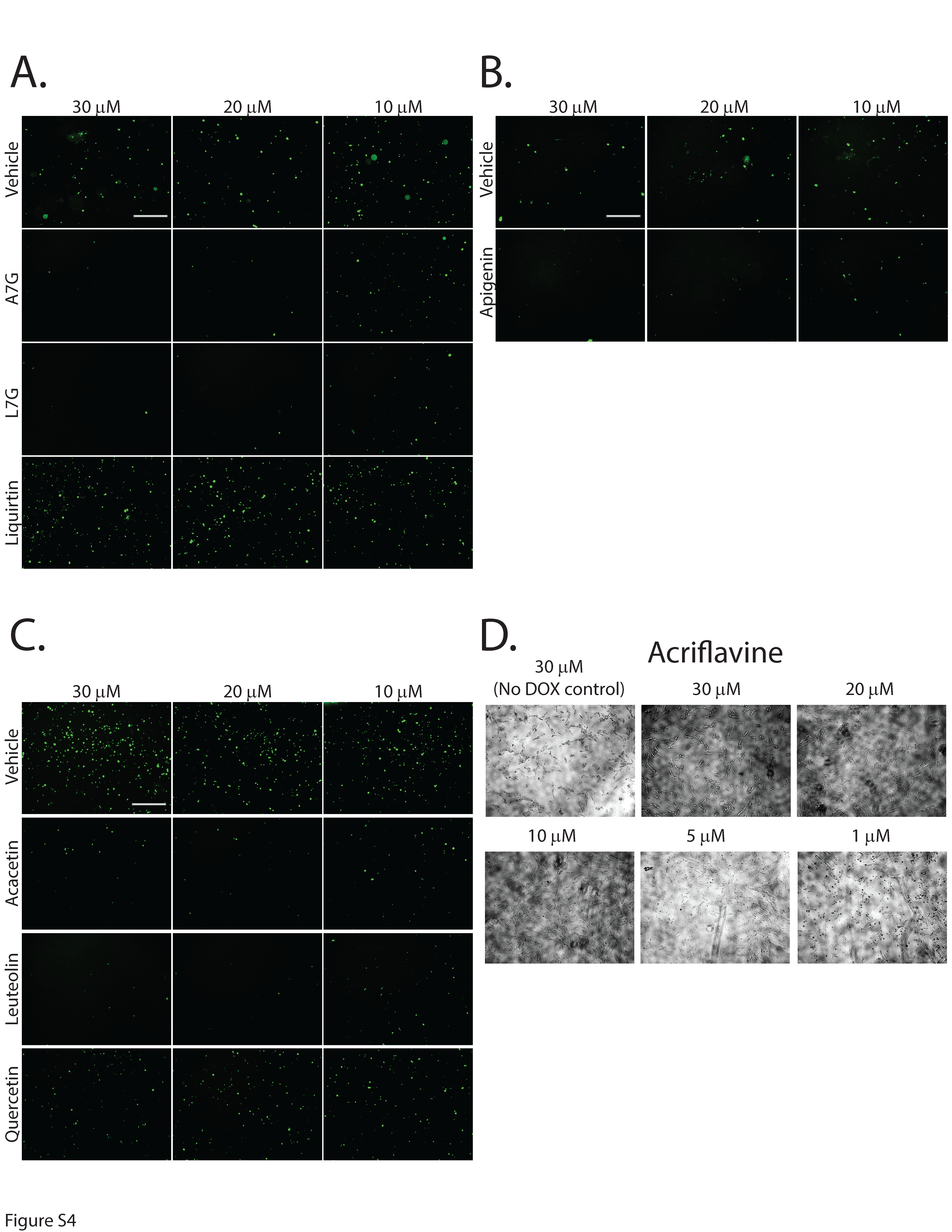
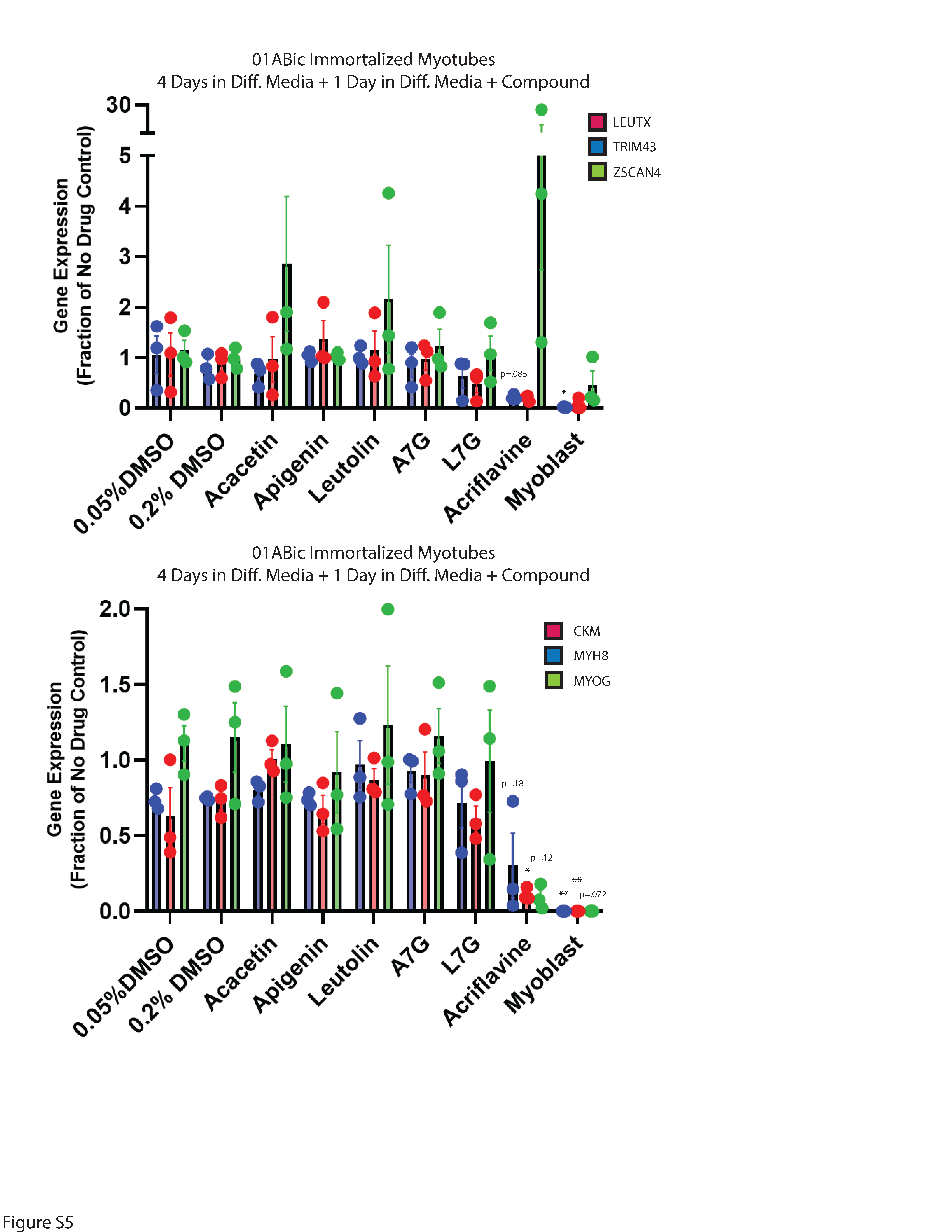
Second-generation small molecules are more potent inhibitors of DUX4-induced apoptosis. While our first-generation compounds were effective at inhibiting DUX4-induced toxicity, the most potent, liquiritigenin, required a 150 mM dose for optimal effectiveness. While this is a significant improvement relative to concentrations required for 4MU, it is still too high for therapeutic use. To overcome this limitation, we performed a second screening of a larger library of compounds (Table 1). Many of these were additional flavone compounds that bear structural similarity to 4MU or to its bioactive metabolite 4MUG (81) (Fig. 3A). For the initial characterization, we used the protocol described above using 50, 5, or 1 mM concentrations and the CellEvent assay (Figure S3). The best performing compounds were then selected for a secondary screening /optimization using concentrations of 30, 20, or 10 mM (Figure S4). This screen identified 5 compounds- acacetin (Cas # 480-44-4), apigenin (Cas # 520-36-5), luteolin (Cas # 491-70-3), apigenin 7-glucoside (A7G, Cas # 578-74-5), and luteolin 7-glucoside (L7G, Cas # 5373-11-5), which provided resistance to DUX4-induced toxicity at optimal concentrations of 20 mM (Fig. 3B). We also identified acriflavine (Cas # 8048-52-0), which based on phase-contrast images provided resistance at 5 mM (Figure S3). We again quantitated these results using the Caspase-Glo assay and found that each compound provided at least four-fold reduction in DUX4-induced toxicity (Fig. 3C). As before, we validated these results using limited-cycle RT-PCR. Acriflavine showed a notable decline in DUX4 transcript levels, suggesting that its effects were an artifact of inhibited transgene activation. Other compounds did not have an effect an DUX4 expression (Fig. 3D). Similarly, western blotting showed that acriflavine treatment significantly reduced DUX4 protein levels, while the remaining compounds did not (Fig. 3E). This again suggests that these compounds exert their effect through a different mechanism than rapamycin. We also examined the effects of these compounds in patient-derived myotubes, and we again found that there was no change in DUX4-target gene expression levels, except for acriflavine, which inhibited expression of all three DUX4 target genes significantly (Fig. 4A). This may have been a result of acriflavine having an effect on myogenesis, as it caused significant overexpression of CKM, while also inhibiting MYOG expression. Similar results were observed using a second patient-derived cell line (Figure S5). These results are again consistent with the five flavone compounds functioning downstream of DUX4 to inhibit toxicity without having negative effects on myogenesis.
Table 1
Second-Generation Compound Screen
|
Compound
|
CAS Number
|
Group
|
|
Acacetin
|
480-44-4
|
4MU-like
|
|
Apigenin
|
520-36-5
|
4MU-like
|
|
Chrysin
|
480-40-0
|
4MU-like
|
|
Farrerol
|
95403-16-0
|
4MU-like
|
|
Fisetin
|
528-48-3
|
4MU-like
|
|
Isorhamnetin
|
480-19-3
|
4MU-like
|
|
Kaempferol
|
520-18-3
|
4MU-like
|
|
Luteolin
|
491-70-3
|
4MU-like
|
|
Myricetin
|
529-44-2
|
4MU-like
|
|
Quercetin
|
117-39-5
|
4MU-like
|
|
Scutellarein
|
529-53-3
|
4MU-like
|
|
4MUG
|
881005-91-0
|
4MUG-like
|
|
Apigenin 7-glucoside (A7G)
|
578-74-5
|
4MUG-like
|
|
Liquiritin
|
551-15-5
|
4MUG-like
|
|
Luteolin 7-glucoside (L7G)
|
5373-11-5
|
4MUG-like
|
|
Myricitrin
|
17912-87-7
|
4MUG-like
|
|
Vitexin
|
3681-93-4
|
4MUG-like
|
|
Scutellarin
|
27740-01-8
|
4MUG-like
|
|
Acriflavine
|
8048-52-0
|
Misc.
|
|
Imatinib
|
152459-95-5
|
Misc.
|
|
Londamine
|
50264-69-2
|
Misc.
|
|
Oxythiamine
|
136-16-3
|
Misc.
|
|
Mitoquinone (MitoQ)
|
845959-50-4
|
Misc.
|
|
PT2385
|
1672665-49-4
|
Misc.
|
|
Shikonin
|
517-89-5
|
Misc.
|
|
TAT-cyclo-CLLFVY
|
1446322-66-2
|
Misc.
|
Flavones and rapamycin inhibit DUX4-induced toxicity through distinct mechanisms. We next investigated the mechanism of action of the flavones. First, we used phospho-specific antibodies to determine how activation of DUX4 affects signaling in the mTor/AKT pathway. We analyzed either uninduced MB135-DUX4i myoblasts, or myoblasts induced with 2 mg/mL doxycycline for 5 or 24 hours with antibodies specific to AKT phosphorylated on Thr308 or Ser473 or phosphorylated ribosomal S6 protein, a marker of mTor activity. After 5 hours of induction, we observed no change in phosphorylation of S6 or AKT at Ser473, and only a small but reproducible increase in AKT phosphorylation at Thr308 (Fig. 5A). However, we observed a notable decline in phosphorylation of AKT after 24 hours of DUX4 expression. In contrast, there was no change in the levels of overall AKT or S6 protein. Thus, it may be that prolonged DUX4 expression inhibits signaling along this axis, or that signaling along the mTor/AKT axis is lost at this late time point, as these populations are actively undergoing apoptosis. To determine the effects of our compounds on AKT/mTor activation, we pre-treated myoblasts with compounds for three hours and then induced DUX4 expression for five hours. As expected, rapamycin and everolimus ablated S6 phosphorylation and triggered hyperphosphorylation of AKT, particularly on Thr308 (Fig. 5B). Surprisingly, of the compounds under study, only honokiol showed a reproducible, but partial inhibition of S6 phosphorylation, and none showed hyperphosphorylation of AKT. Thus, with the possible exception of honokiol, it appears that the first-generation compounds inhibit toxicity through an mTor-independent mechanism. Interestingly, the effects are not DUX4-specific, as performing the same experiments without inducing DUX4 yielded nearly identical results (Fig. 5C), suggesting that they function not by inhibiting a DUX4-activated signal transduction pathway, but rather by triggering a DUX4-independent response that protects against toxicity. To confirm that these observations also hold true for the second-generation compounds, we also pretreated MB135-DUX4i myoblasts with luteolin or L7G for three hours and either left them uninduced for five more hours or induced them with doxycycline for five or 24 hours and analyzed by western blotting (Figure S6). As before, the second-generation compounds showed no inhibition of S6 phosphorylation.
Second-generation compounds prevent loss of ULK1 and induce a marker of autophagy. To investigate the mechanism of action of the second-generation flavone compounds, we considered other pathways that may protect against DUX4-induced toxicity. Luteolin has been previously reported as both a positive and negative regulator of autophagy (82), suggesting that this pathway may be relevant. Also, rapamycin is a known autophagy activator (83), and autophagy regulators integrate multiple signaling pathways (84). Therefore, both mTor-dependent and mTor-independent mechanisms that regulate autophagy may affect DUX4-induced toxicity. We investigated this possibility by analyzing ULK1 expression, a key autophagic regulator that integrates multiple signaling pathways (84). After 5 hours of induction there was no notable change in the levels of ULK1 protein (Fig. 6A). However, after 24 hours much of ULK1 protein was lost. Surprisingly, the decline in ULK1 was accompanied by an increase in LC3-II, a marker of active autophagy. To investigate the effects of the second-generation compounds on autophagy, we pre-treated myoblasts for three hours and then induced DUX4 for 24 hours. We found that the flavone compounds protected ULK1 from DUX4-induced loss. This effect appears to be specific to ULK1 and not a general property of the autophagic machinery, as DUX4 induction did not cause loss of the autophagy-associated proteins ATG3, ATG5, ATG7, or ATG16L1, and the flavones had no effect on these proteins (Figure S7). Interestingly, we observed that each of the flavones increased the abundance of the LC3-II autophagy marker well above the level induced by DUX4 alone (Fig. 6B), suggesting that cellular autophagy protects against DUX4-induced toxicity and that the flavone compounds function by enhancing this protective mechanism. Taken together, these observations identify these flavone compounds as potential drugs for further development, and the autophagy pathway in general as a target for future FSHD therapeutics.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}