Copper-catalyzed selective alkynylation with N-propargyl carboxamides as nucleophiles has been successfully developed for the synthesis of C2-functionalized chromanones. Under optimized reaction conditions, 21 examples were obtained in one-pot procedure through 1,4-conjugate addition. This protocol features readily available feedstocks, easy operations and moderate to good yields, which provides viable access to pharmacologically active C2-functionalized chromanones.
Research Article
Copper-catalyzed C2-selective alkynylation of chromones via 1,4-conjugate addition
https://doi.org/10.21203/rs.3.rs-2453624/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 07 Mar, 2023
You are reading this latest preprint version
Copper-catalyzed
Alkynylation
C2-Functionalized chromanones
1
4-Conjugate addition
One-pot
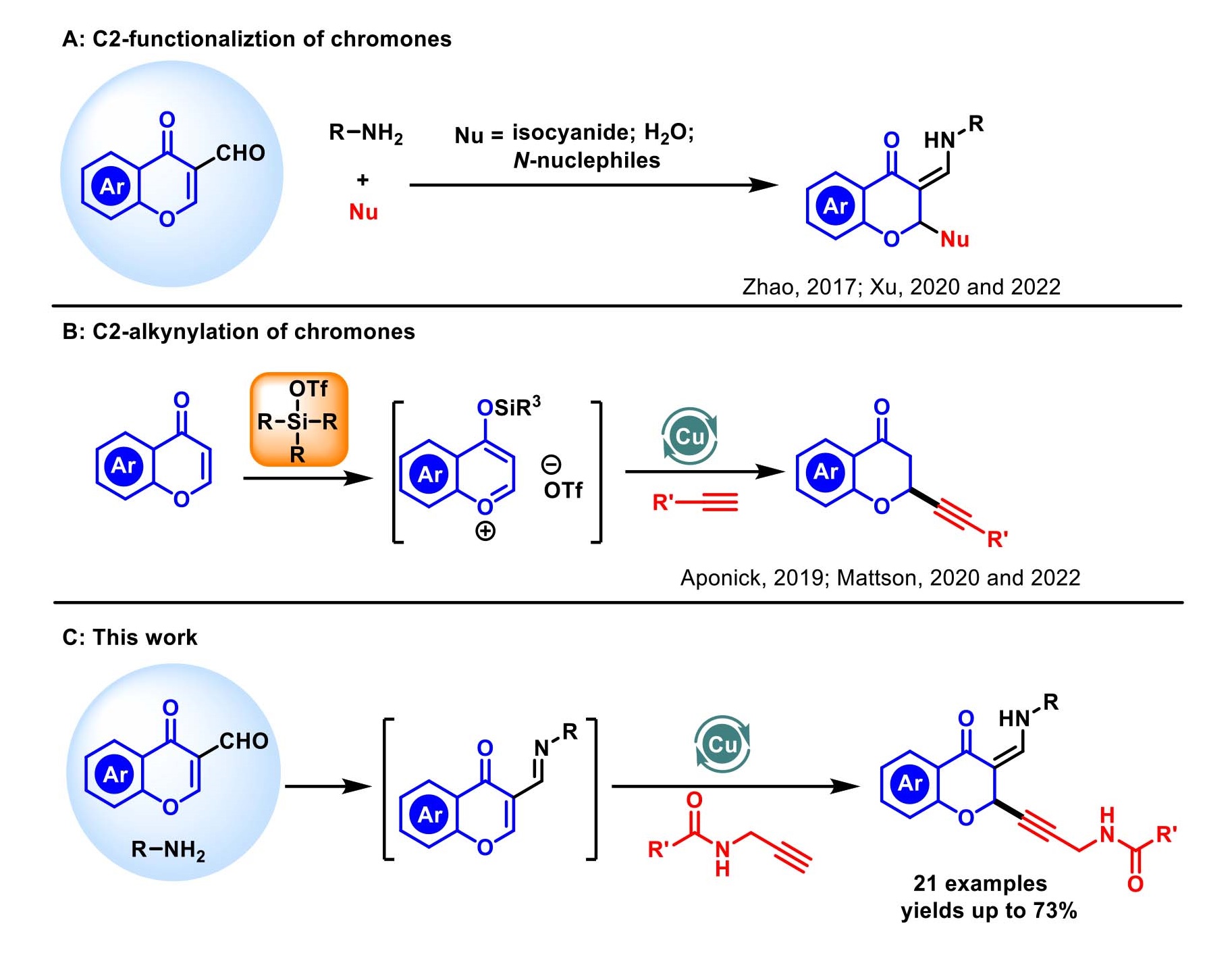
Chromanones constitute a unique class of the core structural motifs in medicinal chemistry and its related 2-functionalized chromanones are widespread throughout bioactive natural products (Fig. 1) [1–8]. Although certain privileged structures display applicability across a range of transformations, the discovery of new methods for the synthesis of these scaffolds is still crucial. Based on the structure-activity relationship (SAR) for chromone, chromone-3-carboxaldehyde was oft-employed synthetic precursors in a panel of high-value transformations, which was proved to be a promising Michael acceptor to rapidly generate diverse chemical libraries with complex form. Thus far, most reports have focused on chromone based-Michael addition by employing strong nucleophiles, such as activated methylenes [9–12], N-nuclephiles [13–18] and isocyanides [19]. Synthesis to valuable 2-pyridone analogues with activated methylenes have been reported by Lee [9], Maiti [10], Ibrahim [11] and Xu [12] through intramolecular- or intermolecular-cascade cyclization of chromones. In an effort to explore the methodologies for pyridine scaffolds, nitroketene-N,S-acetals [13], acetonitriles [14] and amines [15–18] were selected as readily available feedstocks to afford a nitrogen source. Recently, unexpected strategies, employing isocyanides as strong nucleophiles, for 1,4-addition synthesis of chromone analogues were developed by our group, which were controlled by strategically suppressing competing Ugi reaction (Scheme 1A) [20]. To the best of our knowledge, direct chromone-based 1,4-addition of weak nucleophiles has been disclosed but is relatively rare, only water was involved to attack C2 chromones (Scheme 1A) [21]. Despite these important contributions, the implementation of different catalytic transformations of 3-formylchromones to achieve structural diversity in heterocycle synthesis remains an important goal in organic and medicinal chemistry.
Therefore, central to the successful establishment of this chemistry would depend on choosing appropriate nucleophiles. Notably, the addition of aliphatic groups has been reported using basic organometallics [22–23], but these groups do not broadly map onto bioactive compounds [24]. In this regard, the direct addition of alkynes is desirable because they are readily available and diverse synthons. Recently, Aponick and Mattson developed the addition of copper acetylides to benzopyrylium triflates in the presence of ligands (Scheme 1B) [25–28]. So far, many elegant examples demonstrate the applicability of this strategy. However, excess amount of silyl trifluoromethanesulfonate was often employed to generate the key intermediate benzopyrylium triflates in situ. Despite these impressive contributions, more efficient and practical catalytic systems for alkynylation of chromones are still in high demand. Based on our experiences in construction of multicomponent reactions to access biologically active chromanones [20–21], the reactivity at the C2 position is enhanced by the formation of imine. Our group has been interested in learning how to synthesize the C2-selective alkynylation of chromones through an operationally-simplistic. We envisioned that direct 1,4-addition of chromones with alkynes as nucleophiles would be feasible under certain condition, because C2 position of 3-formylchromone was activated by suppressing the A3-coupling reaction [29–30]. Herein, we present a direct copper-catalyzed C2-alkynylation of chromones with N-propargyl carboxamides as nucleophiles through one-pot protocol (Scheme 1C).
We began our investigation for C2-alkynylation with 3-formylchromone 1a, tert-butylamine 2a and N-propargylamides 4a in 2,2,2-trifluoroethanol (TFE) as solvent. As our previous work indicated, Brønsted acids were more capable of generating 1,4-addition adducts under mild conditions. Unfortunately, with these conditions, unsatisfied yield of desired product was observed (Table 1, entry 1 and 2), while the rest was a mixture of the Schiff base 3a and N-propargylamides 4a. Then we turned our attention to Lewis acids, such as ZnCl2, InCl3, FeCl3, CuCl2, Cu(OAc)2, CuSO4 and AgOTf (entries 3–9), CuSO4 led to moderate yield of C2-alkynayl chromanone 5a with dichloroethane (DCE) as solvent. Further screening of various solvents showed that DCE was more suitable for this transformation (entries 10–14). Among the investigation of reaction temperature, acceptable yield of 5a was obtained in 100 ℃ for 3 hours (entries 15–17). Prolonging reaction time to 5 hours produced a dramatic improvement in yield (entries 18–19). So, the optimized reaction conditions were determined to be: “1a (0.3 mmol), 2 (0.3 mmol) and 4a (0.2 mmol) with 0.1 mmol CuSO4 in presence of DCE (2.0 mL) at 100 ℃ for 5 hours.
Table 1. Optimization of reaction conditionsa

|
Entry |
Cat. |
Solvent |
Temp. (℃) |
Time (h) |
Yield (%) |
|---|---|---|---|---|---|
|
1 |
HCOOH |
TFE |
60 |
3 |
< 10 |
|
2 |
HClO4 |
TFE |
60 |
3 |
< 10 |
|
3 |
ZnCl2 |
DCE |
60 |
3 |
16 |
|
4 |
InCl3 |
DCE |
60 |
3 |
21 |
|
5 |
FeCl3 |
DCE |
60 |
3 |
< 10 |
|
6 |
CuCl2 |
DCE |
60 |
3 |
< 10 |
|
7 |
Cu(OAc)2 |
DCE |
60 |
3 |
32 |
|
8 |
CuSO4 |
DCE |
60 |
3 |
47 |
|
9 |
AgOTf |
DCE |
60 |
3 |
NR |
|
10 |
CuSO4 |
MeCN |
60 |
3 |
34 |
|
11 |
CuSO4 |
THF |
60 |
3 |
19 |
|
12 |
CuSO4 |
Toluene |
60 |
3 |
NR |
|
13 |
CuSO4 |
Dioxane |
60 |
3 |
40 |
|
14 |
CuSO4 |
DMF |
60 |
3 |
< 10 |
|
15 |
CuSO4 |
DCE |
80 |
3 |
52 |
|
16 |
CuSO4 |
DCE |
100 |
3 |
61 |
|
17 |
CuSO4 |
DCE |
120 |
3 |
49 |
|
18 |
CuSO4 |
DCE |
100 |
5 |
73 |
|
19 |
CuSO4 |
DCE |
100 |
7 |
65 |
|
aReaction condition: 1a (0.3 mmol), 2 (0.3 mmol), 4a (0.2 mmol), 50 mmol% catalyst, solvent (2.0 mL), in a sealed tube. |
|||||
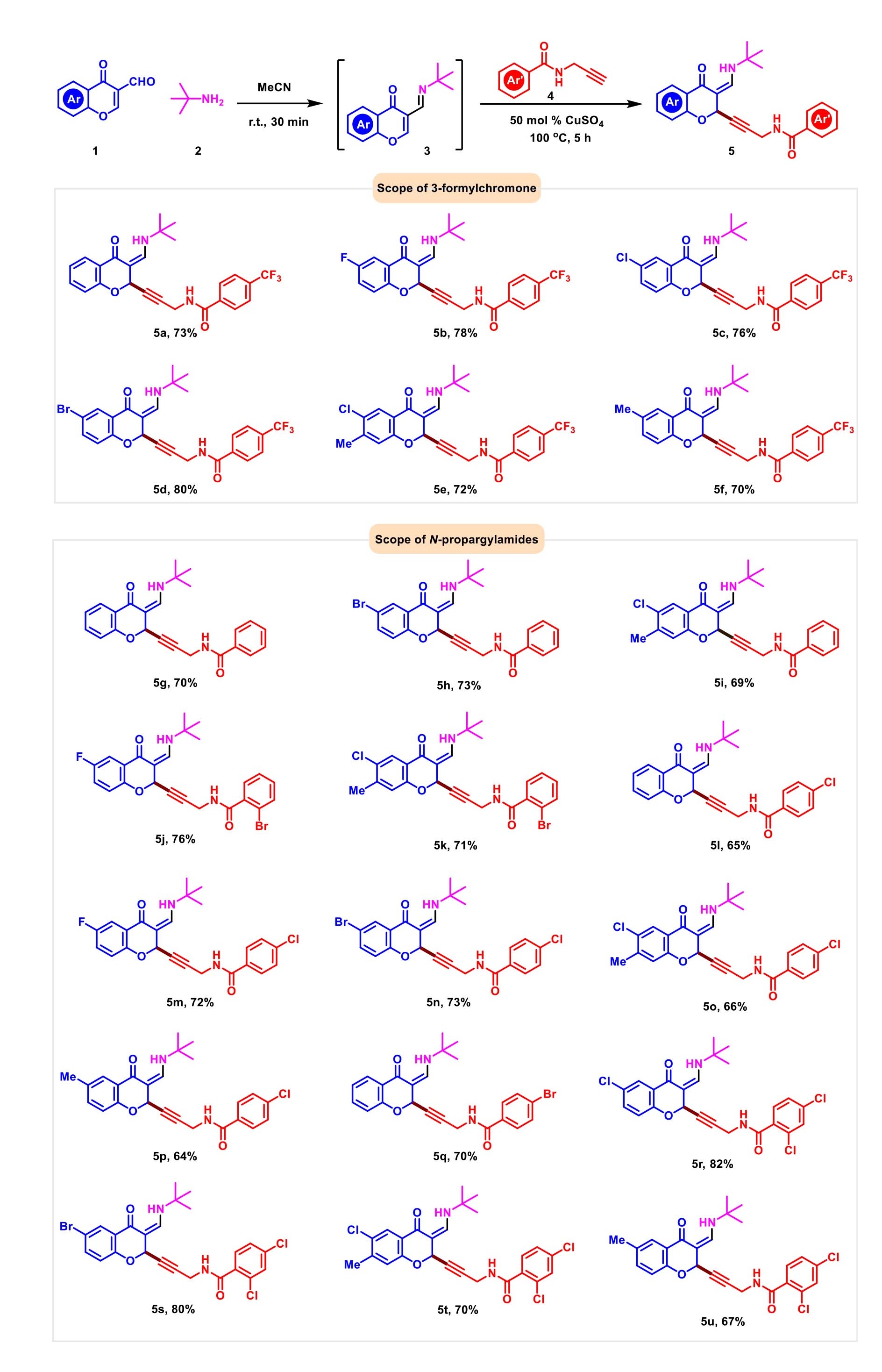
With the optimized conditions in hand, we sequentially evaluated the substrate scope using various chromone-3-carboxaldehydes and N-propargylamides for the generation of a library of C2-alkynyl chromanones (Scheme 2). It was identified that electron withdrawing chromone-3-carboxaldehydes furnished desired products 5b-5e with good yields. To our delight, chromone-3-carboxaldehyde 1 including electron donating group (-Me) also worked well and efficiently afforded 5f with the yield of 70%. For the electron density effect of chromone-3-carboxaldehyde moiety did not dramatically decrease the yields of final products, it suggested that the reaction was robust enough for product conversion. We further examined the scope of N-propargylamides with 50 mol % CuSO4 as catalyst under 100 ℃ for 5 hours. N-Propargylamides containing various groups (H, 2,4-di-Cl, 2-Br, 4-Br and 4-Cl) on the phenyl ring were successful in producing the desired products in good yields (5g-5u). In case that the N-propargylamides with acetyl group did not furnish the desired product, such as N-(prop-2-yn-1-yl) benzamide.
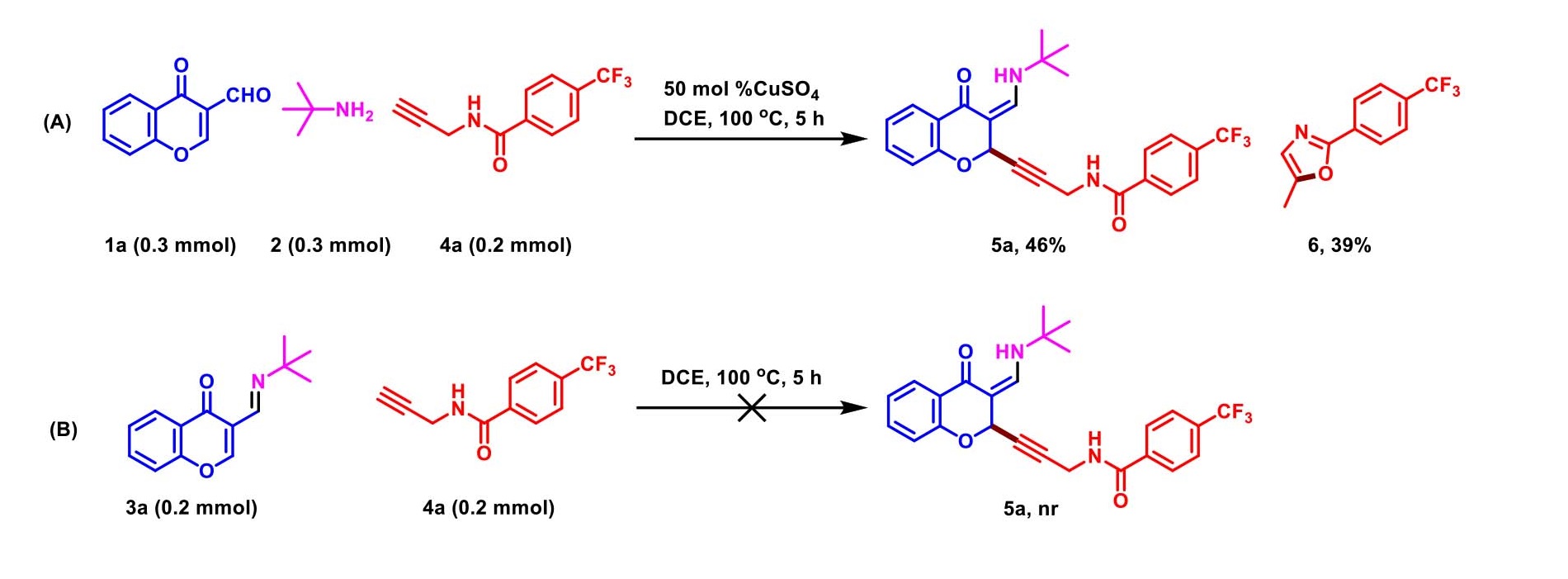
Furthermore, based on previous results [31–32], a series of control experiments was performed as shown in Scheme 3. The mixture of 3-formylchromone 1a, tert-butylamine 2a and N-propargylamides 4a was carried out under standard conditions, diminished yield of 5a was delivered. However, oxazole 6 was also isolated through 5-exo-dig cyclization and aromatic reaction (Scheme 3, A). This indicated that a competitive reaction was involved in C2-selective alkynylation of chromones process, which led to dramatically decrease the reaction yields. Thus, the Shiff base of 3 generated in situ was important for product conversion. Under otherwise identical conditions in the absence of CuSO4, none 2-alkynyl chromanone 5a was observed (Scheme 3, B).
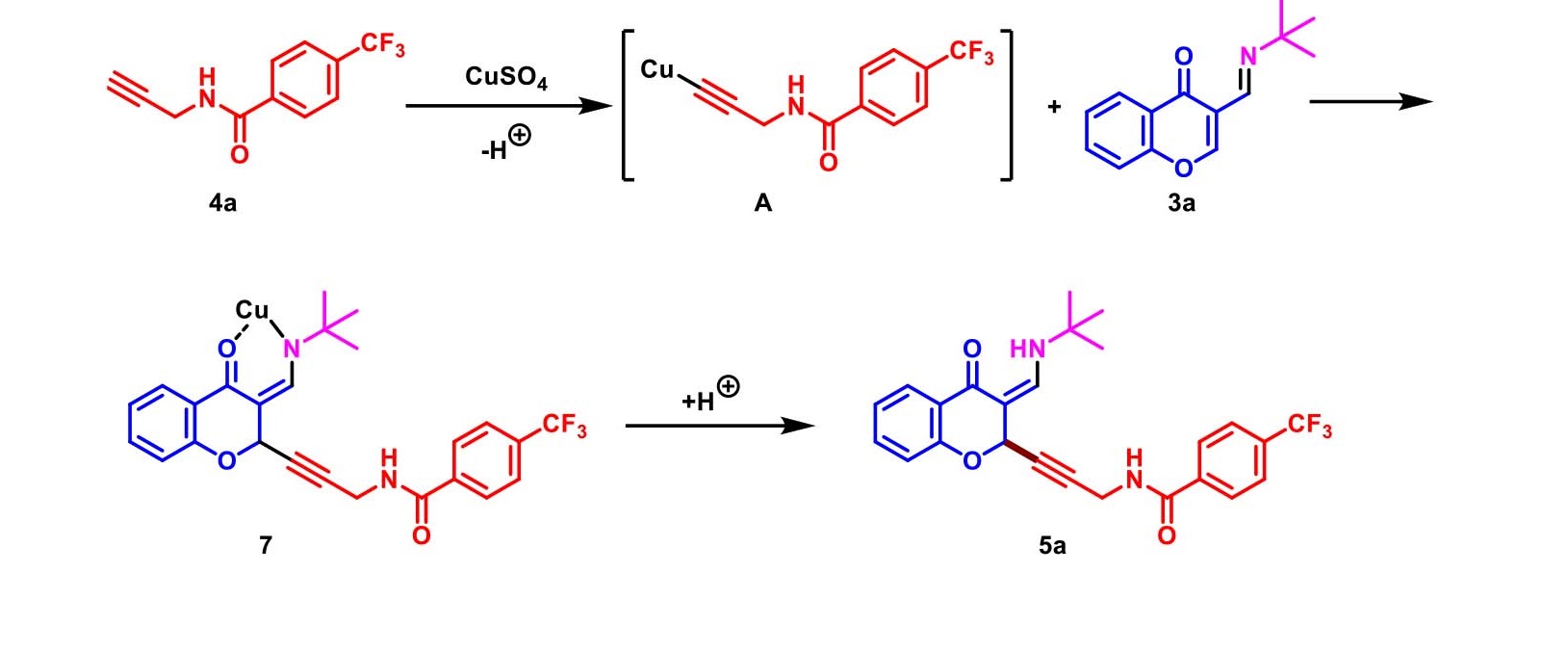
With the above evidence, the proposed reaction mechanism was elucidated as Scheme 4. C2-selective alkynylation of chromones process initiated from formation of copper acetylide A, which was obtained by treatment of CuSO4 with 4a. Then, acetylide attacked to C2-position of 3a to form a complex 7. Finally, protonation gave the desired product 5a.
C2-alkynylation of chromones had been successfully reported by suppressing the A3-coupling reaction. Under optimized reaction conditions, copper-catalyzed 1,4-addition of N-propargyl carboxamides furnished various substituted chromanones with moderate to good yields. This simple protocol provided alternative approach for synthesis of C2-alkynylation of chromanones without pre-activation of chromone. With the evidence of control experiments, the proposed reaction mechanism was presented. Furthermore, MTT assay of synthesized compounds is currently under way in our laboratory.
Acknowledgments
The authors would like to thank the National Natural Science Foundation of China (51973042), Major Project of Natural Science Foundation of Guizhou Province (Grant No. QKHJC-ZK[2021]ZD048), the Science and Technology Research Program of Chongqing Municipal Education Commission (KJQN201801321 and KJQN202101340) the Natural Science Foundation Project of CQ CSTSC (cstc2018jszx-cyzdX0023 and cstc2021jcyj-bsh0233) and Chongqing University of Arts and Sciences: Program for Talents Introduction (R2021FYX05 and P2022YX10) We would also like to thank Ms HZ. Liu for obtaining the LC/MS, HRMS and NMR data.
1H and 13C NMR were recorded on a Bruker 400 spectrometer. 1H NMR data are reported as follows: chemical shift in ppm (δ), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), coupling constant (Hz), relative intensity. 13C NMR data are reported as follows: chemical shift in ppm (δ). LC/MS analyses were performed on a Shimadzu-2020 LC-MS instrument using the following conditions: Shim-pack VP-ODS C18 column (reverse phase, 150 x 4.6 mm); a linear gradient from 10% water and 90% acetonitrile to 75% acetonitrile and 25% water over 6.0 min; flow rate of 0.5 mL/min; UV photodiode array detection from 200 to 400 nm. High-resolution mass spectra (HRMS) were recorded on Thermo Scientific Exactive Plus System. The products were purified by Biotage Isolera™ Spektra Systems and hexane/EtOAc solvent systems. All reagents and solvents were obtained from commercial sources and used without further purification.
General procedures for compounds 5a-u.
A solution of aldehyde (0.3 mmol) and amine (0.3 mmol) in MeCN (1.0 mL) was stirred at room temperature for 30 min in a 5.0 mL microwave vial. Then, N-propargyl carboxamides (0.20 mmol) and CuSO4 (0.1 mmol) were added separately. The mixture was stirred at 100 oC for 5 h. The reaction was monitored by TLC. After the microwave vial was cooled to room temperature, the solvent was removed under reduced pressure and then diluted with EtOAc (15.0 mL), washed with sat. Na2CO3 and brine. The organic layer was dried over MgSO4 and concentrated. The residue was purified by silica gel column chromatography using a gradient of ethyl acetate/hexane (1-100%) to afford targeted 5a-u.
- Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ (2000) Natural productlike combinatorial libraries based on privileged structures. 1. General principles and solid-phase synthesis of benzopyrans. J Am Chem Soc 122(41):9939-9953. https://doi.org/10.1021/ja002033k
- Williams CA, Grayer RJ (2004) Anthocyanins and other flavonoids. Nat Prod Rep 21(4):539-573. https://doi.org/10.1039/B311404J
- Lee H, Lee K, Jung JK, Cho J, Theodorakis EA (2005) Synthesis and evaluation of 6-hydroxy-7-methoxy-4-chromanone- and chroman-2-carboxamides as antioxidants. Bioorg Med Chem Lett 15(11):2745-2748. https://doi.org/10.1016/j.bmcl.2005.03.118
- Fang SH, Lin KN, Huang XQ, Lu YB, Zhang WP, Wei EQ (2012) Nuclear translocation of cysteinyl leukotriene receptor 1 is involved in oxygen-glucose deprivation-induced damage to endothelial cells. Acta Pharmacol Sin 33(12): 1511-1517. https://doi.org/10.1038/aps.2012.101
- Hadida S, Van Goor F, Zhou J, Arumugam V, McCartney J, Hazlewood A, Decker C, Negulescu P, Grootenhuis PD (2014) Discovery of N,-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide (VX-770, ivacaftor), a potent and orally bioavailable CFTR potentiator. J Med Chem 57(23):9776-9795. https://doi.org/10.1021/jm5012808
- Kikuchi H, Isobe M, Sekiya M, Abe Y, Hoshikawa T, Ueda K, Kurata S, Katou Y, Oshima Y (2011) Structures of the dimeric and monomeric chromanones, gonytolides A-C, isolated from the fungus Gonytrichum sp. and their promoting activities of innate immune responses. Org Lett 13(17):4624-4627. https://doi.org/10.1021/ol2018449
- Zhang W, Krohn K, Ullah Z, Florke U, Pescitelli G, Di Bari L, Antus S, Kurtan T, Rheinheimer J, Draeger S, Schulz B (2008) New mono- and dimeric members of the secalonic acid family: blennolides A-G isolated from the fungus Blennoria sp, Chem Eur J 14(16):4913–4923. https://doi.org/10.1002/chem.200800035
- Krohn K, Michel A, Bahramsari R, Floerke U. Aust HJ, Draeger S, Schulz B, Wray V (1996) Biologically active metabolites from fungi 71); aposphaerin A and B two new chroman-4-ones from aposphaeria sp. Nat Prod Lett 8(1):43-48. https://doi.org/10.1080/10575639608043238
- Maezono SMB, Poudel TN, Xia L, Lee YR (2016) A green synthetic approach to synthesizing diverse 2-pyridones for their exceptional UV shielding functions, RSC Adv 6(85):82321–82329. https://doi.org/10.1039/C6RA18661K
- Sengupta T, Gayen KS, Pandit P, Maiti DK (2012) FeCl3·6H2O-Catalyzed intermolecular-cascade cyclization of acetoacetanilide: aldehyde-tuned synthesis to valuable 2-pyridone analogues. Chem Eur J 18(7):1905-1909. https://doi.org/10.1002/chem.201103354
- Ibrahim MA, El-Gohary NM (2018) Domino reactions between 3-(6-methylchromonyl)acrylonitrile and nucleophilic reagents. Tetrahedron 74(4): 512-518. https://doi.org/10.1016/j.tet.2017.12.030
- Lei J, Xu J, Tang D-Y, Shao J-W, Li H, Chen Z-Z, Xu Z-G (2020) A concise and unexpected one-pot methodology for the synthesis of pyrazinone-fused pyridines. Org Chem Front 7(18):2657–2663. https://doi.org/10.1039/D0QO00590H
- Poomathi N, Perumala PT, Ramakrishna S (2017) An efficient and eco-friendly synthesis of 2-pyridones and functionalized azaxanthone frameworks via indium triflate catalyzed domino reaction. Green Chem 19(11):2524-2529. https://doi.org/10.1039/C6GC03440C
- Sultana S, Maezono SMB, Akhtar MS, Shim JJ, Wee YJ, Kim SH, Lee YR (2018) BF3·OEt2-Promoted annulation for substituted 2-arylpyridines as potent UV filters and antibacterial agents. Adv Synth Catal 360(4):751-761. https://doi.org/10.1002/adsc.201701137
- Liao JY, Yap WJ, Wu JE, Wong MW, Zhao Y (2017) Three-component reactions of isocyanoacetates, amines and 3-formylchromones initiated by an unexpected aza-Michael addition. Chem Commun 53(65):9067-9070. https://doi.org/10.1039/C7CC03468G
- Neo AG, López-GarcÍa L, Marcos CF (2014) Allylic amination of Passerini adducts. Application to the selective synthesis of chromone-substituted α-and γ-amino acid peptidic and retropeptidic units. RSC Adv 4(75):40044-40053. https://doi.org/10.1039/C4RA05719H
- Lepitre T, Biannic RL, Othman M, Lawson AM, Daïch A (2017) Metal-free cascade approach toward polysubstituted indolizines from chromone-based michael acceptors. Org Lett 19(8):1978–1981. https://doi.org/10.1021/acs.orglett.7b00309
- Chand K, Prasad S, Tiwari RK, Shirazi AN, Kumar S, Parang K, Sharma SK (2014) Synthesis and evaluation of c-Src kinase inhibitory activity of pyridin-2(1H)-one derivatives. Bioorg Chem 53:75-82. https://doi.org/10.1016/j.bioorg.2014.02.001
- Lei J, Li Y, Xu J, Tang D-Y, Shao J-W, Li H, Chen Z-Z, Xu Z-G (2020) An acid-catalyzed 1,4-addition isocyanide-based multicomponent reaction in neat water, Green Chem 22(12):3716–3720. https://doi.org/10.1039/D0GC00652A
- Lei J, Li Y, He L-J, Luo Y-F, Tang D-Y, Yan W, Lin H-K, Li H, Chen Z-Z, Xu Z-G (2020) Expeditious access of chromone analogues via a Michael addition-driven multicomponent reaction. Org Chem Front 7(8):987–992. https://doi.org/10.1039/D0QO00145G
- Lei J, Ding Y, Zhou H-Y, Gao X-Y, Cao Y-H, Tang D-Y, Li H, Xu Z-G, Chen Z-Z (2022) Practical synthesis of quinolone drugs via a novel TsCl-mediated domino reaction sequence. Green Chem 24(15): 5755–5759. https://doi.org/10.1039/D2GC01689C
- Brown MK, Degrado SJ, Hoveyda AH (2005) Highly Enantioselective Cu-Catalyzed conjugate additions of dialkylzinc reagents to unsaturated furanones and pyranones: preparation of air-stable and catalytically active Cu–peptide complexes. Angew Chem Int Ed 44(33):5306-5310. https://doi.org/10.1002/anie.200501251
- Vila C, Hornillos V, Fañanás-Mastral M, Feringa BL (2013) Catalytic asymmetric conjugate addition of Grignard reagents to chromones. Chem Commun 49(53):5933-5935. https://doi.org/10.1039/C3CC43105C
- Hardman-Baldwin AM, Visco MD, Wieting JM, Stern C, Kondo S, Mattson AE, (2016) Silanediol-catalyzed chromenone functionalization. Org Lett 18(15): 3766-3769. https://doi.org/10.1021/acs.orglett.6b01783
- DeRatt LG, Pappoppula M, Aponick A (2019) A facile enantioselective alkynylation of chromones. Angew Chem Int Ed 58(25):8416-8420. https://doi.org/10.1002/anie.201902405
- Guan Y, Attard JW, Mattson AE, (2020) Copper bis(oxazoline)-catalyzed enantioselective alkynylation of benzopyrylium ions. Chem Eur J 26(8):1742-1747. https://doi.org/10.1002/chem.201904822
- Guan Y, Buivydas TA, Lalisse RF, Attard JW, Ali R, Stern C, Hadad CM, Mattson AE (2021) Robust, enantioselective construction of challenging, Biologically relevant tertiary ether stereocenters, ACS Catal 11(10):6325-6333. https://doi.org/10.1021/acscatal.1c01095
- Guan Y, Buivydas T, Lalisse RF, Ali R, Hadad CM, Mattson AE (2022) Enantioselective dearomative alkynylation of chromanones: ppportunities and obstacles. Synthesis 54(19):4210-4219. https://doi.org/10.1055/a-1811-8075
- Peshkov VA, Pereshivko OP, Van der Eycken EV (2012) A walk around the A3-coupling. Chem Soc Rev 41(10):3790-3807. https://doi.org/10.1039/C2CS15356D
- Rokade BV, Barker J, Guiry PJ (2019) Development of and recent advances in asymmetric A3 coupling. Chem Soc Rev 48(18):4766-4790. https://doi.org/10.1039/c9cs00253g
- Lei J, Li S-Q, Luo Y-F, Tang D-Y, Zhou C-H, Li H, Xu Z-G, Chen Z-Z (2022) Zn(OTf)2‑promoted isocyanide-based three-component reaction: direct access to 2‑oxazolines and β‑amino amides. J Org Chem 87(17):11888-11898. https://doi.org/10.1021/acs.joc.2c01437
- Senadi GC, Hu WP, Hsiao JS, Vandavasi JK, Chen CY, Wang JJ (2012) Facile, selective, and regiocontrolled synthesis of oxazolines and oxazoles mediated by ZnI2 and FeCl3. Org Lett 14(17):4478-4481. https://doi.org/10.1021/ol301980g
Schemes 1 to 4 are available in the Supplemental Files section.
No competing interests reported.
- SI.pdf
- Scheme1.jpg
Scheme 1. C2-functionlization of chromones.
- Scheme2.jpg
Scheme 2.Scope of 2-alkynyl chromanones.
- Scheme3.jpg
Scheme 3. Control experiments.
- Scheme4.jpg
Scheme 4. Plausible mechanism.
{kind=link}
{kind=link}
{kind=link}
{kind=link}