2.1: Isolation of endophytic diazotrophic bacteria
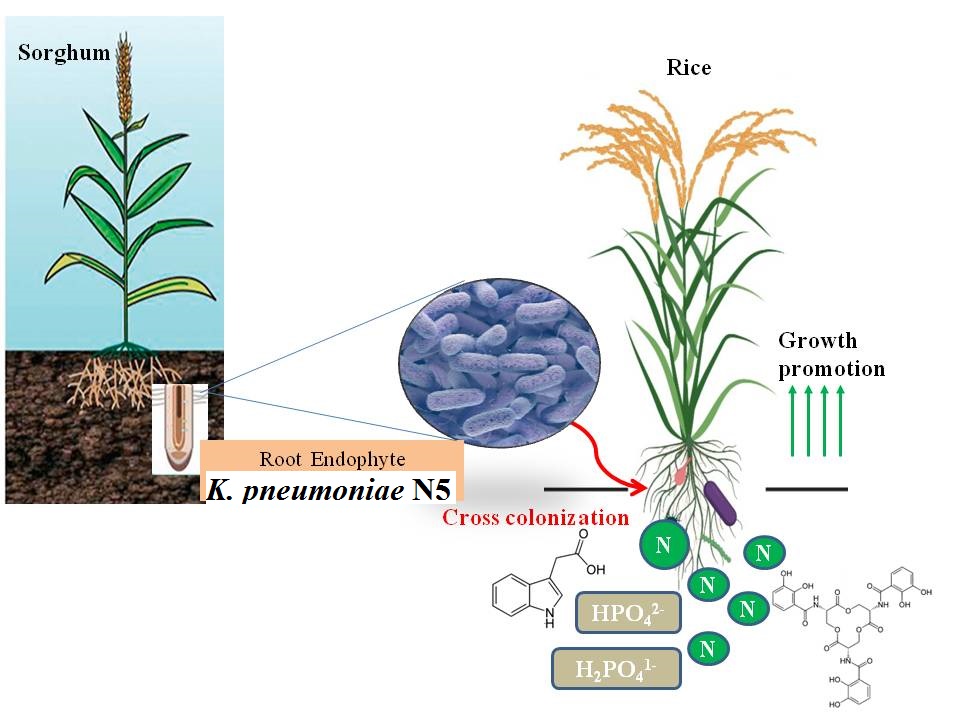
Sorghum plant samples were collected from 5 different regions of South Gujarat, India i.e. Surat, Dandi, Navsari, Valsad and Vapi. Roots of selected plant samples were surface-sterilized with 90% alcohol for 3 min followed by 1% chloramines treatment for 30 min. Treated samples were washed thoroughly with sterile water. Surface-sterilized plant materials were thoroughly crushed in 4% sucrose solution using mortar and pestle and suspension was streaked on Liquid Glucose Ivo (LGI) plates for purification [14]. As a sterility check, 1 ml from the final washing was transferred to 9 ml of LGI broth and 100 µl of this solution was spreaded on LGI plates.
2.2: Screening Of Selected Isolates
To select a suitable isolate having better nitrogen fixing potential, screening of bacterial isolates obtained from 5 different regions, were done based on presence of nifH gene. Amplification of nifH region having size of 360 bp was done using degenerate primer sequences [15]. Pol F primer- TGC GAY CCS AAR GCB GAC TC and Pol R primer-ATS GCC ATC ATY TCR CCG GA. PCR reaction in 50 µl reaction volume was set with: 5 µl of genomic DNA, 5 µl of PCR buffer (10x), 3 µl of MgCl2, 1.25 µl of dNTP mixture, 0.5 µ mol of forward and reverse primers each and 0.2 µl of Taq DNA polymerase. PCR reaction was performed in a thermocycler (eppendorf) and programmed as follows: Initial denaturation at 95oC for 5 min, followed by 35 cycles of denaturation at 94oC for 1 min, annealing at 56oC for 1 min, extension at 72oC for 2 min. PCR amplicon (s) were run on 1% agarose gel and visualized under UV gel documentation system.
2.3: Characterization Of Selected Isolate
Selected bacterial isolate was grown on LGI agar plate at 37oC and pure culture was used for morphological and biochemical characterization as per Bergey‘s Manual of Systematic Bacteriology [16, 17].
2.4: Molecular Characterization Of Endophytic Isolate
The selected endophytic isolate N5 was identified by 16S rRNA gene sequencing. Genomic DNA was extracted by CTAB method [18] and the concentration of total genomic DNA was adjusted to a final concentration of 20 ng µl− 1 for PCR amplification. The 16S rRNA gene of the isolate was amplified using two universal primers namely 27f (5’AGAGTTTGATCCTGGCTCAG3’) and 1492r (5’GGYTACCTTGTTACGACTT3’) [19]. Standard PCR conditions were maintained as: Initial denaturation at 94°C for 5 min followed by denaturation at 94°C for 1 min, annealing at 52°C for 45 sec, and elongation at 72°C for 1 min. At the end of 30 cycles, the final extension step was at 72°C for 8 min [20]. Cycle sequencing reactions were performed using ABI prism terminator cycle sequencing ready reaction kit and electrophoresis of the products were carried out by using AB prism Sequencer, 3130 Genetic analyzer (Applied Bio systems) with 4 capillaries. Further, BLAST was carried out for similarity search of the selected isolate against the GenBank database (website: http://www.ncbi.nih.gov/BLAST).
2.5: Study Of Pgpr Traits
2.5.1: Indole Acetic Acid (IAA) production
K. pneumoniae N5 was inoculated in LB medium (Luria bertani) supplemented with 1 mgml− 1 of tryptophan and incubated at 28oC for 24 h under shaking condition. Further, 2 ml culture of isolate was centrifuged at 1000 rpm for 10 min. Then 2–3 drops of orthophosphoric acid and 4 ml of Salkowski’s reagent were added to the collected Cell Free Supernatants (CFS). The CFS containing test-tubes were kept at room temperature for 20 min. IAA production was indicated by the development of pink colour. Quantification of Indole-3-acetic acid (IAA) was done at 530 nm using spectrophotometer (Dynamica Halo DB-20, Australia) [21].
2.5.2: Siderophore Production
For siderophore production, the culture of isolate was inoculated at the center of plate. Orange purple or dark purplish-red halos around the colonies on blue agar are indicative of siderophore production by selected strain [22].
2.5.3: Phosphorous Solubilisation
Actively growing bacterial culture was spot inoculated on Pikovaskya agar plates with composition (gL− 1): (Yeast extract 0.5, Dextrose 10, (NH4)2SO4 0.5, Ca3(PO4)2 5, KCl 0.2, MgSO4 0.1, MnSO4 0.0001, FeSO4 0.001 and Agar 15, at pH 7.0), and incubated at 30oC for 3 days. Positive results were observed by development of transparent zone against white opaque background.
2.5.4: 1-aminocyclopropane-1-carboxylate (Acc) Deaminase Production
The isolate was point inoculated on salt minimal medium containing ACC as sole nitrogen source. Development of dark red color considered as positive test for ACC deaminase production [23].
2.5.5: Ammonia Production
Bacterial isolate was grown in peptone water for 24 h. 1% inoculum was added to 5 ml of peptone water in each tube and incubated for 72 h at 30oC. Nessler’s reagent (0.5ml) was added in each tube. Development of brown to yellow color indicates positive test for ammonia production [24].
2.6: Estimation Of Nitrogenase Activity
2.6.1: Acetylene reduction assay (ARA)
Nitrogen fixation capacity of nitrogen fixers were quantified indirectly by Acetylene reduction assay (ARA) measuring the reduction of acetylene to ethylene by K. pneumoniae N5 inoculated plants roots [25]. Ten seedlings from each treatment were taken at penicle initiation and grain-filling stages and roots were separated and washed twice with sterile water to remove loosely associated bacteria. The roots were then transferred to fresh, N-free, liquid Jensen’s medium [26]. Test-tubes containing the plant’s root were sealed with a rubber seal and 10% of the headspace volume was replaced with acetylene. Non inoculated plant roots and test-tubes not injected with acetylene served as a controls. They were returned to growth chamber and incubated in the dark for 12 h at 30oC. Acetylene reductase activity was determined using a Gas Chromatography (Shimadzu GC-14A with Porapak-N 80/100 – INOX column).
2.6.2: Endophytic Treatment Of Rice Seed
Healthy seeds of variety Jaya were obtained from main rice research centre, Navsari Agricultural University, and washed thoroughly with distilled water followed by surface sterilization using 0.1% HgCl2 solution for 4 min and 70% ethanol for 10 min. Then seeds were washed thoroughly with sterile distilled water. Surface sterilized seeds were coated with bacterial culture by incubation for 3 h followed by drying. Culture coated seeds were sown in pot and maintained under green house. All recommended practices and plant protection measures were adopted to obtain healthy plants [27]. The observations were recorded viz, root length, shoot length, fresh weight of root and shoot, dry weight of root and shoot, chlorophyll content, leaf area, nitrogen content from plant and soil after 60 days of inoculation.
2.6.3: Scanning Electron Microscopy (Sem)
Plant tissues collected 7 dpi were washed twice with sterile water, fixed with 2.5% (v/v) glutaraldehyde and 4% (v/v) paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.2–7.4, for 2 h at 28◦C. Fixed tissues were washed three times with phosphate buffered saline (PBS, pH 7.2) and cut to separate roots and aerial parts. Plant segments were post-fixed in 1% (w/v) osmium tetroxide in PBS, for 2 h at 4◦C, dehydrated in a 50–100% (v/v) gradient ethanol series, and dried by the CO2 critical point method in a Balzers apparatus, model CDP-20. Samples were subsequently mounted on aluminum stubs with double coated carbon conductive tape (Pelco Int.) and sputtered with gold in a Balzers apparatus, model FL-964. Observations and micrographies were made in a Jeol JSM-5310 scanning electron microscope.
2.7: Statistical Analysis
Mean and standard deviation were calculated for all experiments. Each experiment was conducted in three replications. Data were analyzed by standard analysis of variance (ANNOVA) using Duncan`s multiple range test (DMRT) by Statistic Analysis System (SAS 9.1).
{kind=link}