Most of what it is currently known about the clinical characteristics of ALS comes from studies in white-Caucasians from Europe and North America. However, recent works have shown that epidemiology and clinical manifestations of this disease vary in relation to ethnicity of different populations (4–6). Despite this, only few investigations have evaluated the phenotype of ALS in patients from Latin American countries (11–13). In this context, our results provide a valuable description of the neurological characteristics and survival of Hispanic ALS patients from Mexico City. Notably, we found certain particularities in the phenotype of our population compared to what it is described in the literature. Firstly, the mean age at disease onset in the current study was lower than reported in Caucasian series from Europe (4,14–18), Japan (19), and Israel (20), but it coincides with values observed in the United States (21), Brazil (22), and Uruguay (11). Nonetheless, a previous investigation performed in the northern region of Mexico showed that the mean age at disease onset was around 47 years (13), much lower than observed in the current study. These discrepancies may reflect differences in the genetic background of ALS patients from distant regions around the world, and even from different places within the same country. Indeed, it is now widely recognized that some genetic abnormalities account for specific clinical phenotypes in ALS, especially among patients with the familiar form of the disease. Such genetic alterations may influence the progression of the disease as well as some clinical characteristics such as the age at symptom onset (3). Unfortunately, our study participants were not subjected to any genetic analysis, thus we were unable to address possible genotype-phenotype relationships accounting for differences in the clinical phenotype of our population. Future studies integrating genetic data would provide an estimation of the genetic burden in Hispanic ALS patients and a better understanding of genetic factors affecting their clinical phenotype.
Secondly, we also found a higher incidence of ALS in women, and an increased percentage of bulbar ALS cases, which differs from most of previous reports [4, 6–19]. This increased proportion of bulbar onset ALS patients observed in our study could be ascribed to possible misdiagnosis of spinal onset cases, which are frequently confounded with other syndromes affecting the spinal cord and the peripheric nervous system, such as lumbar stenosis, polyneuropathy, spinal cord tumours, among other (24). Meanwhile, the proportion of familial ALS in our population was about 5%, as described in the global literature (4).
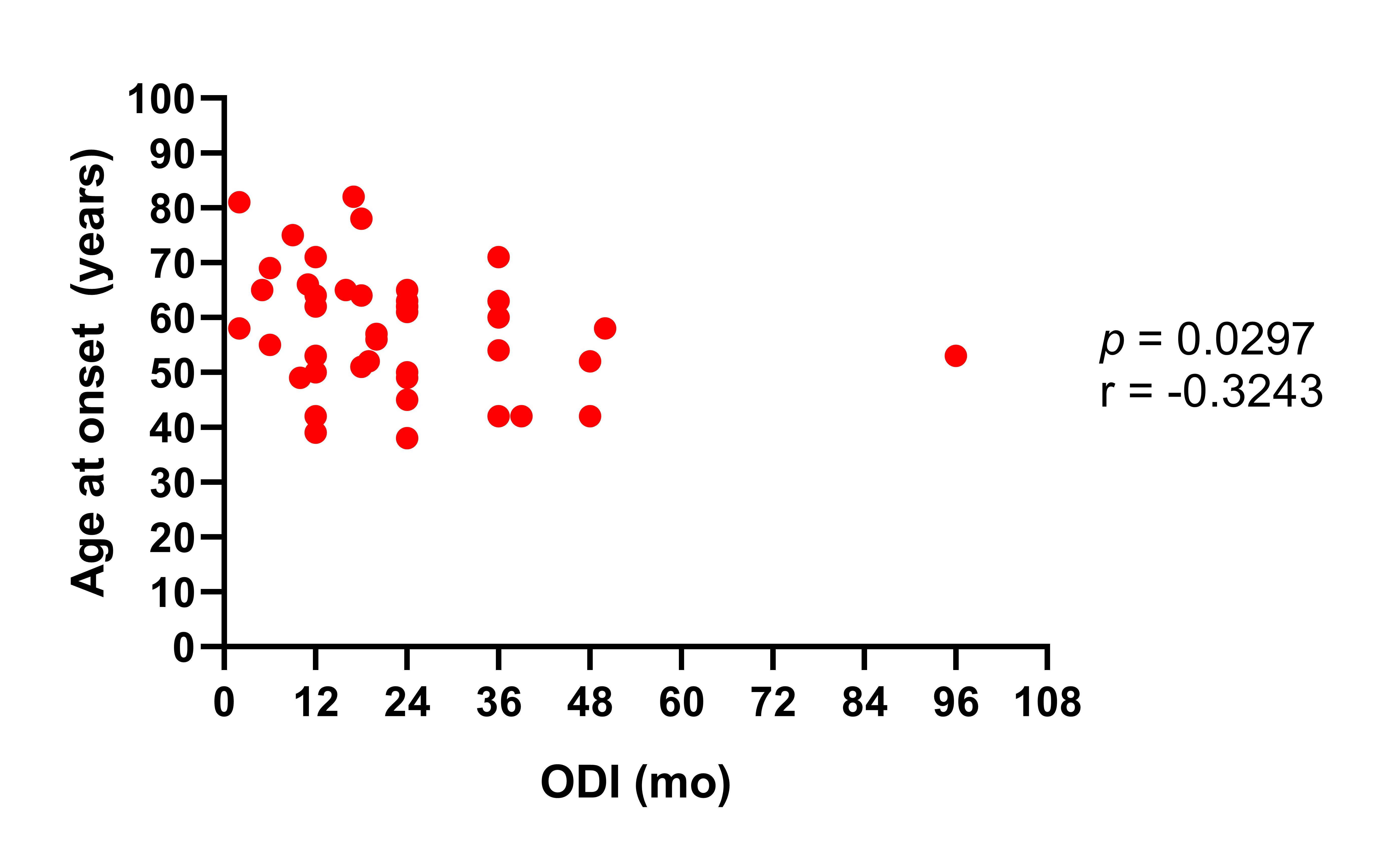
We also addressed the effect of different clinical factors on the prognosis of ALS patients. Of note, the mean survival time observed in our population is one the longest reported in the literature (4), together with survival rates showed in another study carried out in a different region of our country (13). As any of our study participants received treatment with riluzole, the survival rates showed here were not influence by any disease-modifying therapy. Hence, it is possible that the differences in survival rates observed between our patients and other populations may be explained at least in part by variations in the presentation of certain predictive clinical characteristics, such as the age at disease onset and the anatomical region of symptom initiation. In this regard, we found that participants of the current study had a younger age at disease onset, which is a predictor of better survival in ALS (4). In fact, our results showed that age at onset was inversely correlated with survival time in our population. Nonetheless, as we mentioned before, we also observed a higher proportion of ALS patients presenting with the bulbar onset form of the disease, which has been related to poor survival in previous studies as well as in the current report (4).
On the other hand, genetic factors influencing the progression of the neurological decline in ALS patients, together with environmental exposures may account for discrepancies in survival among different ethnic groups, as mentioned before. Furthermore, survival can also be influenced by differences in the nutritional support, respiratory management and other medical interventions provided to ALS patients in distinct regions. For instance, variations in the management of respiratory failure in ALS are observed between centres located in different continents (25). Such management differences may also exist between distinct types of hospitals located in the same region. In this context, the survival rates observed in the current study may not represent the actual life expectancy of ALS patients from different regions of our country, as our data were obtained from a single third level medical centre where there might be a better access for ALS patients to get specialized management for their disease complications compared to other hospital around Mexico. Regarding other clinical predictors of survival in ALS, our results confirmed that the need for PEG and MV during the course of the disease is a predictor of poor outcome in our patients, whereas gender did not affected the overall survival of our ALS patients, contrary to other studies showing a slightly higher mortality among women (19,22,23). Similarly, body weight reduction after diagnosis, a well-recognized independent predictor of mortality in ALS (26), had non predictive significance in our study.
As it has been reported in previous studies (13,21,23,27), our data demonstrated that a longer diagnosis delay after onset of symptoms is a robust protective factor for mortality in ALS cases. Interestingly, we also found that patients with shorter intervals between symptom onset and diagnosis were older than individuals with longer diagnostic delay, which may explain our observations, as it is well known that there is a significantly better survival in younger ALS patients (18,21,23,27). This means that ALS patients that begin with symptoms at older ages seek medical attention earlier, but as they may have a lower capacity to compensate for the declining in motor functioning and a higher burden of other comorbidities, their life expectancy is shorter and then, their shorter interval between disease onset and diagnosis predicts a poor survival. This phenomenon may also be related to variations in the rate of disease progression among patients looking for medical attention early after disease onset compared to those attending late to their first clinical examination. In fact, Czaplinksi and colleagues have shown that the more rapidly a patient initially deteriorates the shorter the diagnostic delay (21), suggesting that patients with a faster progressing of disease seek medical care earlier, whereas those with slower disease progression attend to their first examination later as they can compensate better for their initial symptoms. Apart from this, the time of diagnosis delay reported here doubles the value observed in most of the studies from Europe and the United States (4), which may reflect limitations in the diagnostic approach to ALS patients in our settings.
Finally, some studies have previously reported that the concentrations of lipids in the blood of patients with ALS measured at the time of diagnosis have a positive impact on their survival (9,10). Specifically, Dorst and colleagues showed that high fasting serum levels of cholesterol at baseline predicted a longer survival in a group of ALS patients (10). Similarly, Dupuis and colleagues demonstrated that steatosis of the liver was more pronounced in ALS patients compared to individuals with Parkinson disease, and that elevated cholesterol levels increased the survival of ALS patients by more than 1 year (9). Conversely, other studies have shown that the lipid profile has no prognostic value in ALS (28,29), although lower serum lipids have been found to be related to respiratory impairment in ALS patients (28). In this context, our data may contribute to clarify the role of lipids in ALS, as we confirmed that higher serum levels of total cholesterol at diagnosis are associated with longer survival and patients underwent to both MV and PEG have lower levels of total cholesterol compared to individuals who did not require such interventions. Collectively, these data suggest that a greater energy reserve in the form of lipids may delay the process of motor neuron degeneration and muscle atrophy in ALS patients, who have shown an elevated resting energy expenditure (30).
Our study possesses all the restrictions of a retrospective design in relation to the access to clinical information. For instance, we could not retrieve additional data about the time of dysphagia onset in all the participants to address the impact of such variable on their survival, thus we only reported the time at which PEG was required. Similarly, forced vital capacity (FVC) was not regularly monitored in all the participants, thus we could not evaluate the predictive value of FVC at baseline nor the impact of any decrease in FVC on survival in our population (31). Furthermore, although the primary outcome in our study was survival, we were unable to include other measurements of disease severity such as the ALS functional rating scale revised (ALSFRS-R) score to address clinical characteristics predicting functional decline in our patients (32). Similarly, the fact that the current study was conducted at a single third-level medical centre made us unable to perform a population-based comparison, which limited our ability to estimate the local incidence of ALS and may restrict the representativeness of our data. Moreover, a better description of the ethnic background of the participants would have provided useful information for future investigations aimed to compare clinical factors and prognosis of ALS patients across populations with different genetic ancestry. Finally, a major limitation of our study is that any participant was subjected to genetic studies to identify some of the genetic abnormalities known to be associated with familiar and sporadic ALS as acknowledged before (3). In this regard, although clear genotype–phenotype relationships where the presence of a specific mutation accurately predicts clinical characteristics do not exist, a better genetic characterization of our population would have provided relevant information to identify possible mutations accounting for differences in certain characteristics of the phenotype of our patients, for instance, their longer survival and their earlier age at disease onset. Despite these limitations, our study represents one of the few clinical descriptions of Hispanic ALS patients available in the literature, providing evidence of a specific phenotype in our population.
{kind=link}