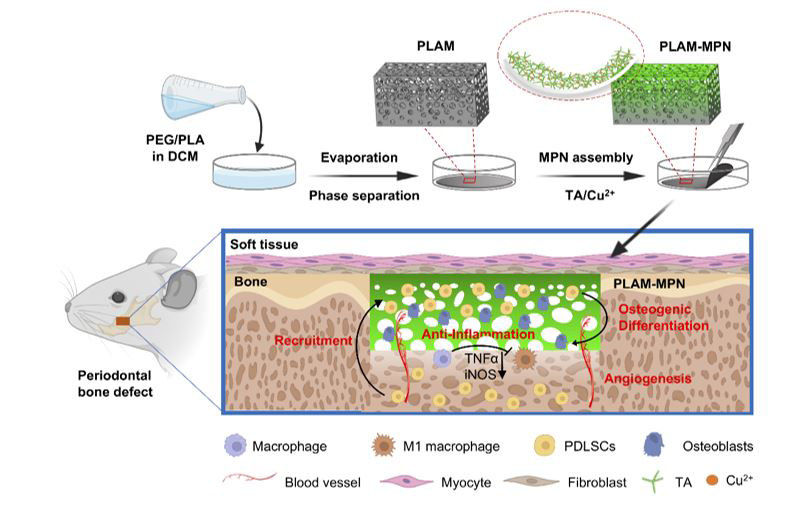
2.1. Fabrication of the scaffolds
PLAM were prepared by evaporative phase separation method. The preparation process was as follows: dichloromethane (DCM) was added to a glass bottle, and then polyethylene glycol (PEG) 200 was added to DCM, the mass ratio of PEG and DCM was 1:5. The mixture was vigorously stirred, then PLA (13.3% w/w DCM) was dispersed in the PEG solution, and the glass vial was sealed and placed under sonication at 37 ℃ to minimize solvent evaporation. The sonication was supplemented with stirring until the mixture was homogeneous. The mixture was poured over a glass dish to distribute evenly and the glass dish was then placed in a fume hood at room temperature for 24 h to remove residual solvent. Finally, by removing the thin film (300-500 µm in thickness) from the glass plate, the Janus porous barrier film was obtained.
2.2. Fabrication of the MPN
MPN was obtained by a layer-by-layer method. The preparation process was as follows: the prepared PLAM scaffold in the plate was washed three times with deionized water. Next, the PLAM scaffold (100 mm in diameter) was soaked with 10 mL of TA solution (0.8 mg mL−1) at room temperature, shaken for 30 s at 75 rpm, followed by the addition of 10 mL of Cu2+ solution (0.2 mg mL−1), shaken again for 30 s at 75 rpm. Then, 100 μL of sodium hydroxide solution (pH=13) was added to adjust the pH to 7.5 and shaken for 30 s at 75 rpm. Finally, the scaffold was washed thrice with deionized water to obtain the MPN-loaded PLAM scaffold. According to the number of MPN layers required subsequently, the above preparation process was repeated 1, 5 and 10 times to obtain PLAM-1MPN (PLAM-1), PLAM-5MPN (PLAM-5) and PLAM-10MPN (PLAM-10) scaffolds, respectively.
2.3. Characterization of the scaffolds
The morphology of the scaffolds was observed using a scanning electron microscope (SEM) (Pro G5, Phenome) at an accelerating voltage of 5 kV. To corroborate the different chemical composition of the PLAM and the PLAM-MPN scaffolds, ultraviolet spectrophotometer (UV) (Shimadzu, UV2600 Ι) was used to detect the characteristic peak of Cu-TA. To detect whether Cu2+ was loaded successfully on the surface of the PLAM-MPN scaffolds, Fourier infrared spectrometer (FTIR) (Bruker, ALPHA Ⅱ) was used to scan over the range of 50-3500 cm−1.
The surface wettability of the scaffolds was detected by static water contact angle measurement at room temperature (26±1 ℃). A contact angle instrument (Zhongchen, JC2000) was utilized, where 2.0 µL deionized water was automatically dropped onto the scaffold and recorded using a digital camera until the water droplet on the scaffold displayed a stable shape. Three points at least from different positions of each sample were recorded and averaged to determine the static water contact angle.
The tensile strength of scaffolds (10 × 50 mm) was characterized using a universal materials tester (Shimadzu, Japan). All samples were stretched at a constant tensile rate of 10 mm min−1 and the original length of the strips was 30 mm. Each sample was measured for three times in the tensile tests.
2.4. Cell culture
The following study protocol was approved by the Medical Ethical Committee of School of Stomatology, Shandong University (Protocol Number: GD20210609). The isolation of BMDMs was carried out based on previous procedures 23.The BMDMs were cultured in DMEM medium contained 20% fetal bovine serum (FBS) (BioInd, Kibbutz, Israel) and 1% penicillin/streptomycin and 20 ng mL−1 of macrophage colony-stimulating factor (M-CSF) (Protein-tech, Chicago, USA). Fresh medium was changed regularly until 80-90% confluent monolayers were obtained.
The following study protocol was approved by the Medical Ethical Committee of School of Stomatology, Shandong University (Protocol Number: GR20210323). The isolation of human PDLSCs was carried out based on our previously reported procedures 24. Briefly, human PDLSCs were cultured in DMEM supplemented with 10% FBS. PDLSCs at passages 4-6 were used for the following experiments. HUVECs were commercially purchased (ScienCell, San Diego, USA) and seeded in supplemented endothelial culture medium (ScienCell). All cells were cultured at 37 ℃ in a 5% CO2 incubator with a humidified atmosphere.
The scaffolds were cut into 24 mm in diameter for 6 -well culture plate or 14 mm for 24 -well plate. The scaffold membranes were soaked in 75% ethanol for 2 h, after that these scaffolds were rinsed with PBS solution to remove residual ethanol. Then, the scaffolds were dried under sterile conditions and soaked in α-MEM for 24 h. BMDMs, PDLSCs and HUVECs were seeded on top of the scaffolds respectively for subsequent experiments.
2.5. Cell viability evaluation
The cytotoxicity of the PLAM-MPN and pure PLAM scaffolds was examined by a LIVE/DEAD viability/cytotoxicity kit (Invitrogen, CA, USA). PDLSCs were seeded at a density of 5×104 cells/well and incubated for 1, 2 and 3 d. At the end of each time period, the medium was discarded. The scaffolds were rinsed gently thrice with PBS. Next, the staining working solution were prepared according to the instructions: 5 μL calcein-am solution and 10 μL propidium iodide (PI) solution were respectively added to 5 mL PBS. Finally, the different samples were observed under the microscope and photographed. Cell viability and proliferation on different substrates were evaluated by CCK-8 (Dojindo Laboratories, Tokyo, Japan) after cultivated for 1, 2 and 3 d.
2.6. PDLSC morphology on the scaffolds
First, the growth state of PDLSCs on the scaffolds was observed by confocal laser scanning microscopy (CLSM, LSM880). In order to identify the porous structure of the scaffold, the red rhodamine B fluorescent dye was added to the PLAM-B, PLAM-P, PLAM-1, PLAM-5, PLAM-10 surface. All the following experimental operations were performed in the dark. After 48 h culture, the medium was discarded. The scaffolds were rinsed gently thrice with PBS. Then, cells were fixed with 4% paraformaldehyde for 10 min at room temperature. After washing with PBS, the cells were stained with DAPI for 5 min. Finally, after washing again with PBS, the growth state and distribution of cells in the stained porous scaffolds were observed under CLSM. The morphology of PDLSCs on PLAM-10 scaffold was characterized under SEM after dehydration by gradient alcohol.
2.7. In vitro immunoregulation assay
The identity of the obtained BMDMs was tested first by flow cytometry (Accuri-C6, BD Biosciences, San Diego, USA). The detected maker included the F4/80 (17-5920-82, Invitrogen) and CD11b (141708, Biolegend). After being passaged, the BMDMs were seeded onto the barrier membrane at a density of 2×105 cells/well. After culturing for 12 h, the conditioned medium was replaced. Among them, in all groups to detect the M1 polarization, the medium was DMEM containing 100 ng mL-1 LPS + 20 ng mL-1 IFN-γ in 10% FBS, and the stimulation time was 12 h. The groups to detect the M2 polarization, except that the medium of the "NC+IL-4" group was 20 ng mL-1 IL-4+20 ng mL-1 IL-10 in DMEM with 10% FBS, the rest medium in the group was DMEM with 10% FBS alone, and the stimulation time was 48 h. Next, phenotypic characterization of BMDMs induced by the different scaffolds was performed by flow cytometry. Stimulated BMDMs were incubated with allophycocyanin (APC)-conjugated anti-iNOS (2102823, Invitrogen) and anti-CD206 (MMR) (141707, Biolegend).
The gene expression levels of tumor necrosis factor (TNF-ɑ), inducible nitric oxide synthase (iNOS), arginase-1 (Arg-1), and CD206 were measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) to assess the immunoregulation effects of the PLAM-MPN scaffolds on BMDMs, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primer was selected as the housekeeping gene and the sequences of the primers were provided in Supplementary Table S1.
In addition, for fluorescence analysis, the samples were fixed with 4% paraformaldehyde for 10 min, and then 10% goat serum was added and incubated at 37 ℃ for 1 h. Subsequently, the cells were incubated with iNOS antibody (1:300, ab49999, Abcam) and CD206 antibody (1:300, ab64693, Abcam) overnight, respectively. The cells were further incubated with CoraLite 488-conjugated goat anti-rabbit secondary antibody (1:800, SA00013-2, Protein-tech) and then counterstained with DAPI (ab104139, Abcam) for 5 min. The images were captured with the fluorescence microscope (Olympus).
2.8. In vitro vascularization assay
After the HUVECs were cultured with different scaffolds respectively for 14 d, the tubule formation assay was performed according to the manufacturer's instructions. The Matrigel were placed at 4 ℃ overnight and thaw and the 48-well culture plate and pipette tips were pre-cooled at -20 ℃ for 1 h in advance. Then, the thawed Matrigel was spread evenly on the pre-cooled 48-well plate (100 μL/well), placed at 37 ℃ for 30 min to solidify the Matrigel. While waiting for Matrigel to solidify, cells grown on negative control (NC), PLAM, and PLAM-MPN scaffolds were collected by trypsinization and washed twice with PBS. The cells were then resuspended at a density of 5.5×105 mL−1. Finally, 100 μL of cell suspension was added to each well of a 48-well plate. After 8 h of incubation, the samples were observed and photographed under microscope. The relative genes (vascular endothelial growth factor (VEGF), stem cell factor (SCF) and hypoxia-inducible factor (HIF)) expression levels of angiogenic differentiation were analyzed by qRT-PCR. GAPDH primer was selected as the housekeeping gene and the sequences of the primers were provided in Supplementary Table S1.
2.9. In vitro stem cell recruitment assay
The PDLSCs were digested and adjusted to 5×105 mL−1 in α-MEM supplemented with 0.1% FBS, and 200 μL of the cell suspension were inoculated to the upper side of the migration chamber. The scaffolds were then placed in the lower side and soaked in 500 μL of α-MEM containing 0.1% FBS. The NC group was also cultured in α-MEM with 0.1% FBS, and the PC group was cultured with α-MEM with 10% FBS. After the cells incubated at 37 ℃ for 20 h, the noninvasive cells on the upper side were removed by a cotton swab. Finally, the cells were fixed, stained with crystal violet and observed under an optical microscope (Olympus, Tokyo, Japan).
2.10. In vitro PDLSC osteogenesis assay
The PDLSCs were seeded at a density of 1.5×105 cells/well. After the cell adhesion, the medium was replaced with osteogenic induction medium. After 21 d, the samples were rinsed gently with PBS to avoid wrapping the cells on the culture plate. Then the cells were fixed with 4% paraformaldehyde for 30 min. After being rinsed, an appropriate amount of Alizarin Red staining solution was added to each well for 30 min. A digital camera was used to collect the optical image of the overall macroscopic effect of the orifice plate, and the microscopic image of the mineralized nodules was observed under a microscope and photographed. Next, 1 mL of 10% cetylpyridinium chloride (CPC) solution was added to each well of the 6-well plate. After the mineralized nodules were dissolved for 30 min at room temperature, 100 μL of the solution were pipetted into a 96-well plate. Finally, the OD value was measured and analyzed at 562 nm. The relative genes (alkaline phosphatase (ALP), runt-related transcription factor 2 (Runx2) and osteopontin (OPN)) expression levels of osteogenic differentiation were analyzed by qRT-PCR. GAPDH primer was selected as the housekeeping gene and the sequences of the primers were provided in Supplementary Table S1.
2.11. In vivo periodontal bone regeneration assay
Animal studies were approved by the Ethics Committee of Stomatological Hospital of Shandong University (Protocol Number: GD20190901). The animal experimental protocols were performed according to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. Wistar rats (male, 7 w, 220±20 g, Chales River, Beijing, China) were used in this study (24 rats for each group, 72 rats in total). they were stored in the animal center at the Shandong University following a 12+12 hours dark/light cycle with a fasten 20-25°C ambient temperature, food (provided and water were autoclave-sterilized prior to administration. The mice were free to access to food and water prior and after experiments.
According to the previous published articles 24,25, the operation was performed under pentobarbital sodium anesthesia (40 mg kg−1 body weight). In brief, bilateral periodontal bone defects (5×4×1 mm3, L×H×D) were created with a dental drill, and the defect area was located approximately 1 mm posterior to the anterior border of the mandible and 1 mm below the superior border of the mandible. All rats with defects were numbered and randomly divided into 3 groups, in which different scaffolds were implanted into the defects: (1) no treatment (NC), (2) PLAM, and (3) PLAM-10. After surgery, the animals were intramuscularly injected with penicillin (400000 U mL−1) for three successive days. At 1, 2, 4 and 8 w, the rats were sacrificed by excessive pentobarbital anesthesia, then the mandible specimens of the rats were collected and fixed with 4% paraformaldehyde by cardiac perfusion for the following experiments.
2.12. Micro-computed tomography assay
To estimate and compare bone regeneration of defect site in each group, micro-computed tomography (Micro-CT) (PerkinElmer, MA, USA) analysis was applied. Quantitatively, the parameters including the bone volume/total volume (BV/TV),bone surface/total volume (BS/TV), trabecular bone thickness (Tb.Th), trabecular bone separation (Tb.Sp) were analyzed and calculated to assess bone regeneration. The implanted scaffolds were taken out at 8 w post-surgery, and the cell growth on the both surfaces of the barrier membrane was observed by SEM.
2.13. Immunofluorescence assay
Decalcification of the specimens was performed with 10% disodium ethylenediaminetetraacetic acid (EDTA-Na2, Solarbio Beijing, China) at 4 ℃ for 30 d. Then, decalcified tissue embedded into paraffin wax was sliced into 5 μm sections. Immunofluorescent staining was performed as described before. In order to detect the bone regeneration, phenotype switching of macrophages and angiogenesis in vivo, the tissue sections were incubated with primary antibodies anti-ALP (1:500, ab65834, Abcam), anti-Runx2 (1:500, ab76956, Abcam), anti-CD68 (1:250, ab955, Abcam)/anti-iNOS (1:200, ab15323, Abcam), anti-CD68/anti-CD206(1:200, ab64693, Abcam) and anti-CD31 (1:300, ab28364, Abcam). The fluorescent slides were finally covered by the cover glass using mounting medium containing DAPI. The fluorescent images were obtained with the fluorescence microscope, the fluorescence intensity of ALP and the number of Runx2+ cells, CD206+/CD68+ cells, iNOS+/CD68+ cells and CD31+ cells in the defects were counted and measured by Image J software.
2.14. Statistical analysis
All values are reported as the mean ± standard deviation (SD) and were analyzed by Student’s t test and one-way ANOVA using GraphPad Prism software (Version 6, MacKiev Software, Boston, MA, USA). Differences were considered statistically significant at P < 0.05.
{kind=link}