HiPSC culture
HiPSC lines were obtained from Bioneer A/S Hørsholm Denmark and University of Copenhagen (UCPH) Copenhagen Denmark (BIONi010-C, BIONi010-C-29, BIONi010-C-30) and Allen Institute for Cell Science (AICS-0016). They were cultured in StemFlex™ medium (Gibco™) with 1 % Penicillin/Streptomycin on Geltrex™ (Gibco™, 1:150 dilution in DMEM/F12) coated 35 mm dishes at 37 °C in 5 % CO2 atmosphere. The parafilm-sealed dishes were stored at 4 °C for up to 2 weeks and were incubated for 60 min at 37 °C prior to use for polymerization. A light microscope was used to observe cell growth and health. The StemFlex™ medium was changed every 1-2 days and the cells were split before 70-80 % confluency was reached. For splitting, the cells were rinsed with 1 ml DPBS (Gibco™) once and harvested by adding 1 ml ReLeSR™ (Stem Cell) for 30 s at RT. The solution was removed except for a thin film and incubated for additional 90 s, occasionally tapping the dish gently on the bench. The undifferentiated cell colonies were detached by rinsing 3-4 times with 1 ml StemFlex™ and divided on new culture dishes.
Gene editing of HiPSC
Before 70 % confluence was reached, TrypLE™ (Gibco™) was used to detach the cells according to the manufacturer’s instructions. The cells were counted using a Neubauer chamber and 200.000 cells per nucleofection sample were prepared according to the manufacturer’s instruction using a Nucleocuvette (Lonza) and P3 primary cell nucleofection buffer (Lonza). Nucleofection was performed according to the instructions of IDT with Nucleofection enhancer, RNP complex (Cas9 with tracrRNA for LMNB1 gene) (IDT) and 400 ng of LMNB1_mTagRFP-T HDR plasmid (Addgene 114403). Nucleofection was done with the pulses DN-100, CA-137 and control (no pulse) with the Lonza 4D nucleofector. HiPSCs were cultivated with 0.1 % ROCK inhibitor (Stem Cell) and 0.1 % HDR enhancer at 37 ºC and 5 % CO2 and the medium was changed 24 h after nucleofection. The fluorescent signal of the knock-in (KI) of the cells was observed by fluorescence microscopy 72 h after nucleofection. Several days post nucleofection, cells were detached using TrypLE™ (Gibco™) as described before and centrifuged. The pellet was resuspended in DPBS in a concentration below 100.000 cells/ml. Cells that successfully included the RFP fluorescent KI were separated by fluorescent-assisted cell sorting (FACS) using the MoFlo Astrios Eq Cell Sorter (Beckman Coulter). More than 3000 cells were sorted in one well of a 96-well plate pre-coated with Geltrex™ and equilibrated with 100 µl Stemflex™ and 0.1 µl ROCK inhibitor. Cells were further incubated at 37 ºC and 5 % CO2 and cultured changing medium every other day. The ACTB-GFP hiPSC line is commercially available and was purchased from Coriell Institute (AICS-0016 hiPSC).
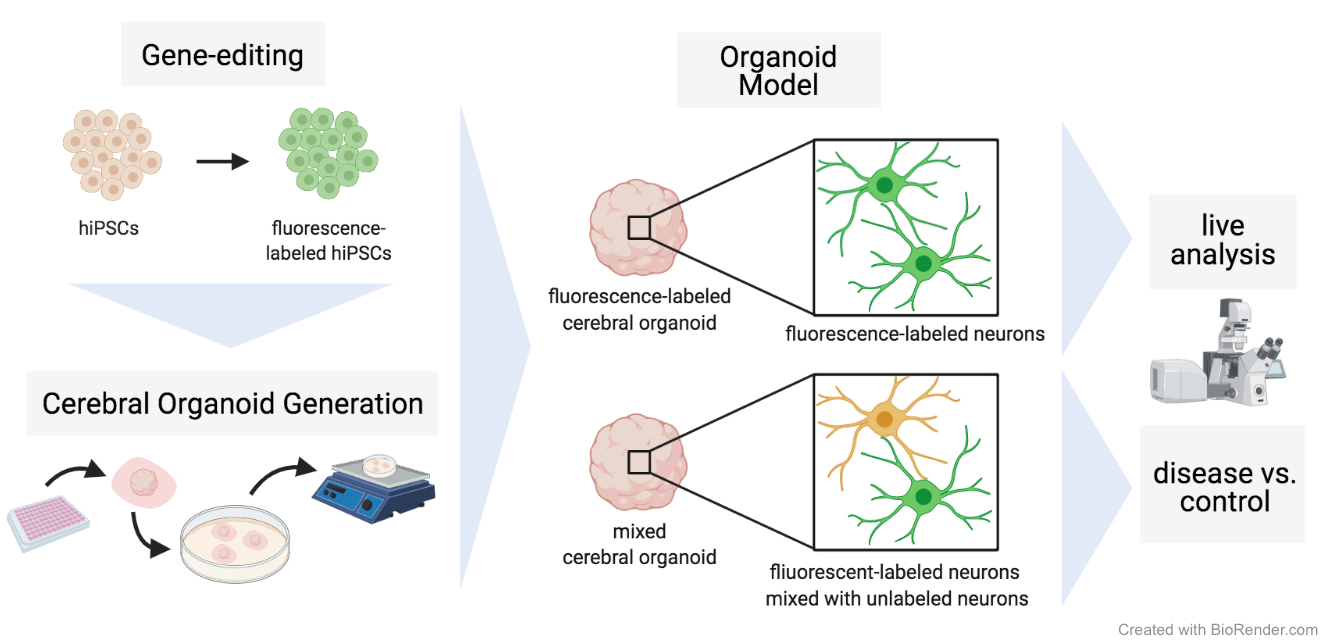
Normal and Mixed Cerebral Organoids
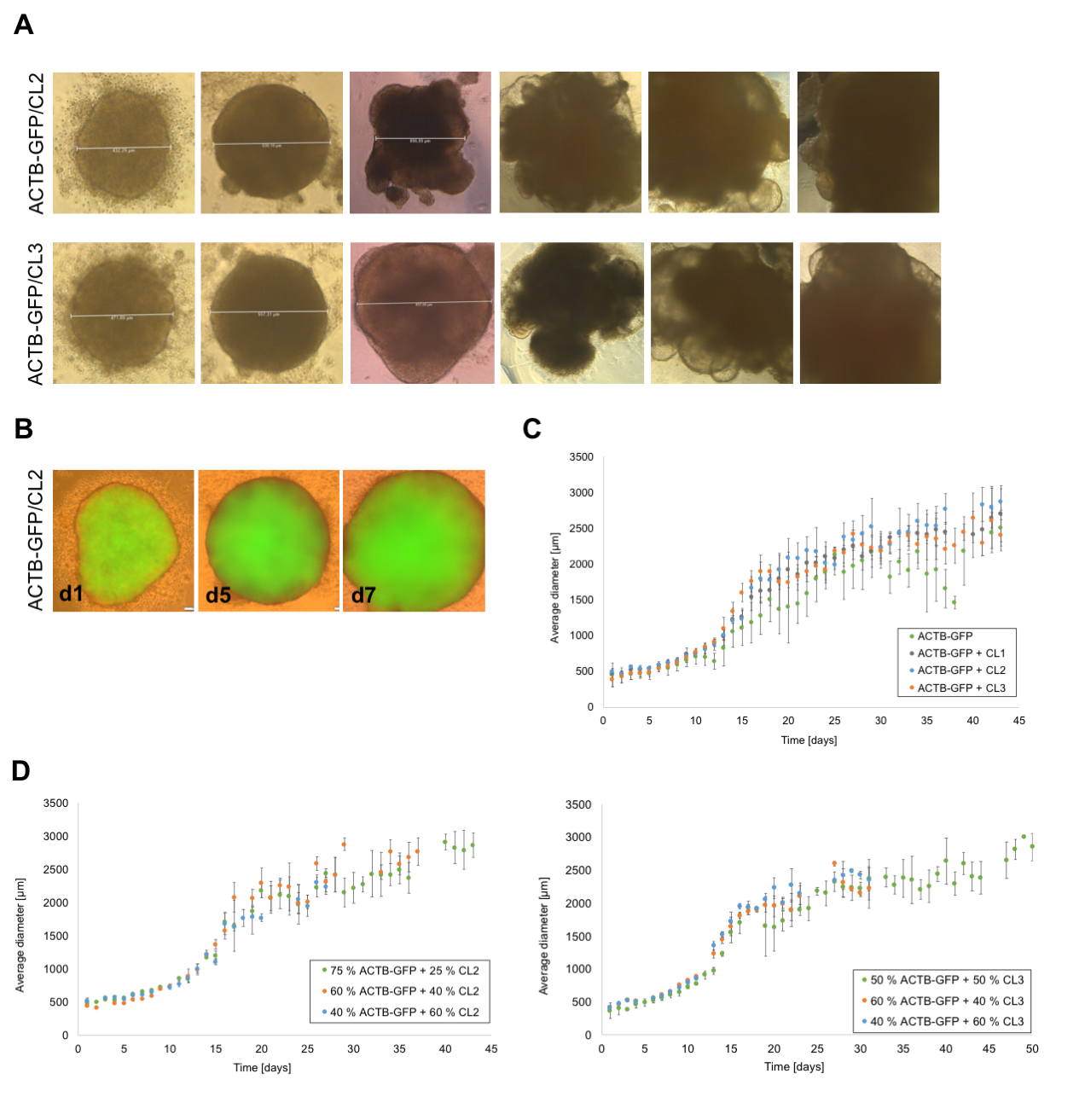
Cerebral organoids were cultured according to the protocol of Lancaster and Knoblich [1] and only modifications are stated in the following. HiPSCs were harvested using TrypLE™ and hiPSC lines (ACTB-GFP, RFP-LMNB1, CL1, CL2 and CL3) were mixed in respective ratios before centrifugation at 300 x g for 5 min at RT. On day 1 and 2 after seeding, the EBs were monitored for their appearance and size. If the EBs showed blurred edges or a diameter smaller than 300 µm, half of the medium was aspirated and exchanged with 150 µl of fresh hESC medium containing 4 ng/ml bFGF and 50 µM ROCK inhibitor. On day 3, the medium was changed to hESC medium without supplements. After six days (day 6), the medium was changed to NI medium and then changed every day until day 11 or 12, when EBs were embedded in growth-factor reduced Matrigel (Corning) droplets. Therefore, the EBs were removed from the well using a cut 200 µl pipette tip and placed in a trough on a parafilm strip. Excess media around the EBs was removed and a 10 µl drop of Matrigel was added to each EB which was then carefully moved to the middle of the drop using a 10 µl pipette tip. After 25-30 min of incubation at 37 °C to allow polymerization of the Matrigel, the Matrigel drops were carefully loosened at the edges using a 10 µl pipette tip and the parafilm strip was placed upside down into a well of a six-well plate filled with 2 ml of DM-A medium. Gentle shaking of the plate detached the Matrigel drops from the parafilm strip and the parafilm strip could be removed from the well. Embedded EBs were kept at 37 °C and 5 % CO2 in a humidified atmosphere.
Organoid cryo- and vibratome-sections and immunofluorescence staining
COs were fixated in 4 % PFA (4 °C, 90 min) and subsequently washed and incubated in a 30 % sucrose solution (4 °C, overnight). The next day, the COs were embedded in a 1:1 mixture of 30 % sucrose solution and Tissue freezing medium (Sakura), snap-frozen on dry ice, and stored at -80 °C. They were sliced into 16 µm sections using a cryostat (Leica CM3050S), mounted on Superfrost™ slides (Thermo Fisher), and stored at -80 °C until further use. For immunofluorescence staining, the following steps were performed at RT and protected from light and washing was done with PBS for 4 min, if not stated otherwise. The CO sections were thawed in PBS and circled with a Liquid Blocker Super PAP pen (Thermo Fisher). After washing twice, the sections were blocked and permeabilized with 0.1 % Triton-X-100 and 5 % goat serum in PBS for 1 h. Primary antibodies were diluted in 0.1 % Triton X-100 in PBS (β -tubulin III: ab78078, abcam, 1:500; MAP2: M4403, Sigma, 1:500; PAX6: 60094, StemCell, 1:500) and incubated on the CO sections in a humidified chamber at 4 °C overnight. The sections were washed twice and incubated with the (biotinylated) secondary antibody, diluted in 0.1 % Triton-X-100 in PBS, in a humidified chamber for 1 h. After washing twice, the sections were incubated with Avidin-TRITC diluted 1:1000 in 0.1 % Triton X-100 in PBS. In case of not using a biotinylated antibody, the secondary antibody was diluted in 0.1 % Triton X-100 in PBS and incubated on the sections in a humidified chamber for 45 min. The sections were washed two times and incubated with 0.001 mg/ml DAPI or 0.001 mg/ml Hoechst33342 for 15 min. Following two washing steps, the sections were mounted with a coverslip and left for drying overnight.
Vibratome slices were generated according to the protocol by Lancaster et al. [11] with some modifications. Briefly, mature organoids (day 35-45) were washed (PBS without Ca2+ and Mg2+) and embedded in 3 % low-melting-point agarose (Thermo Fisher Scientific) at 40 °C in reusable silicone molds (1 cm³). The agarose blocks were cooled on ice for 10 min and sectioned into 300 µm thick slides, using the Leica VT1000S vibrating microtome in cold PBS. Sections were collected in 24-well plates, containing serum-supplemented slice culture medium (SSSCM: DMEM (Thermo Fisher Scientific), 10 % FBS, 0.5 % (w/v) glucose, 1x (v/v) GlutaMAX (Thermo Fisher Scientific), 1 % Antibiotic-Antimycotic (Thermo Fisher Scientific) and were incubated for 1 h at 37 °C. After incubation, the medium was replaced with serum-free slice culture medium (SFSCM: Neurobasal (Thermo Fisher Scientific), 1:50 (v/v) B-27 supplement (Thermo Fisher Scientific), 0,5 % (w/v) glucose, 1x (v/v) GlutaMAX (Thermo Fisher Scientific) and 1 % Antibiotic-Antimycotic (Thermo Fisher Scientific). Sections were cultivated at 37 °C, 5 % CO2 and the medium was exchanged every other day.
Widefield and confocal microscopy
Developing fluorescently labeled EBs were regularly imaged using an Olympus IX51 inverted microscope equipped with a 10x objective. Immunohistochemically stained CO sections were imaged with a Leica TCS SP8 confocal microscope system using a 10x dry objective (0.3 NA) or 100x oil objective (1.4 NA) and a 405 nm and white light laser with excitation wavelengths of 405 nm (DAPI/Hoechst), 488 nm (EGFP) and 551 nm (TRITC) and emission wavelengths of 461 nm (DAPI/Hoechst), 509 nm (EGFP) and 576 nm (TRITC). The detection ranges were 410-463 nm (DAPI/Hoechst), 493-592 nm (EGFP) and 556-701 nm (TRITC). Images were recorded in a sequential scan at a scan speed of 600 Hz into 5648 x 5648 (10x) or 2640 x 2640 (100x) images. Hybrid detectors (HyD) were used for EGFP/TRITC and a photomultiplier tube detector (PMT) for DAPI/Hoechst. For tile scans, 465 x 465 µm areas (3 x 3 tiles) were recorded in x- and y-direction. The LAS X-software was used to analyze the data, and to improve resolution, a deconvolution process was performed via HyVolution 2 in the Huygens Essential Automatic approach, taking the refractive index of the mounting medium into account.
{kind=link}
{kind=link}