LCA patients
A family with a 5-year-old proband (III-2) and two suspected patients (III-1 and III-3) were recruited for this study. Written informed consent was obtained from each individual to participate in this study. The proband was diagnosed with LCA at Guangdong Zhongshan Hospital, Shanghai General Hospital and the Second Affiliated Hospital of the School of Medicine, Zhejiang University. The pedigree was constructed for the proband based on information provided by the guardians.
DNA isolation and qualification
Total genomic DNA was extracted using the Relax Gene Blood DNA System (Tiangen, Beijing, China) following the manufacturer’s instructions. All DNA was dissolved in sterilized double-distilled water and kept at –20 °C until assayed.
One percent agarose gels were used to monitor DNA degradation and contamination. All DNA samples were examined for protein contamination (as indicated by the A260/A280 ratio) and reagent contamination (indicated by the A260/A230 ratio) with a NanoDrop ND 1000 spectrophotometer (NanoDrop, Wilmington, DE, USA).
Targeted-Next generation sequencing (NGS)
DNA samples obtained from the proband were sequenced using targeted-NGS. A customized Sequence Capture 2.1M Human Array from Roche NimbleGen (NimbleGen, Madison, USA) was designed to capture 3093 exons (including 100 bp regions that flanked the exons) from 222 genes known to be associated with common genetic diseases, including retinitis pigmentosa, Waardenburg syndrome, X-linked juvenile retinoschisis, crystalline retinitis pigmentosa, albinism, LCA, Bardet Biedl syndrome and cone-rod dystrophy (Additional Table 1). The procedure for the preparation of the libraries was consistent with standard protocols published previously [8]. The data of targeted-NGS assay was analyzed by Illumina basecalling Software 1.7.
Mutation validation by Sanger sequencing
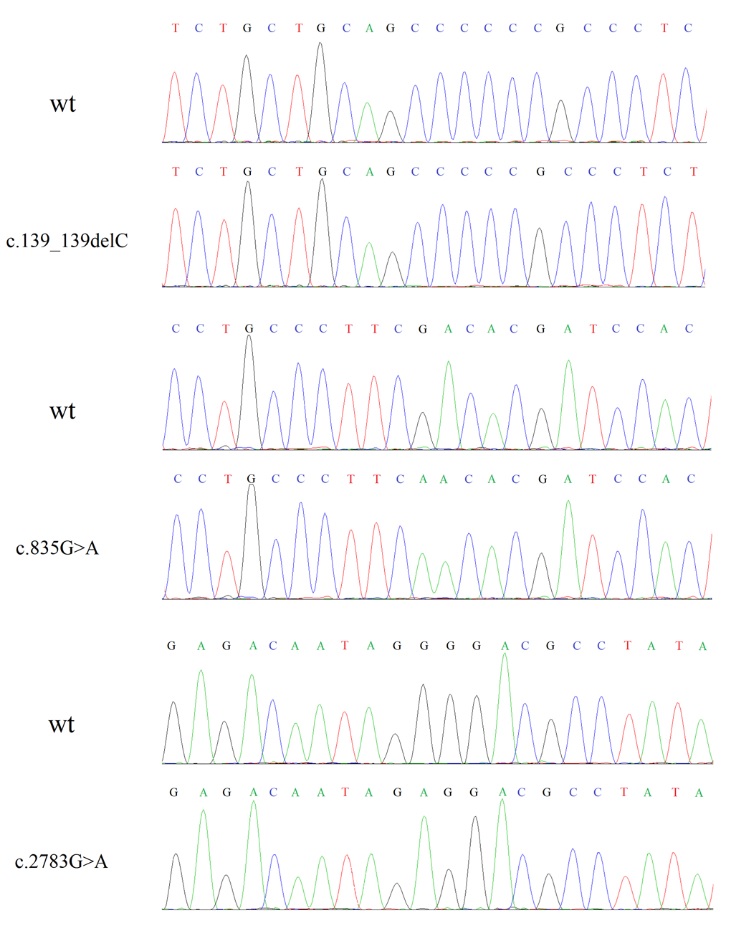
Sanger sequencing was used to validate candidate variants identified by NGS. Primers of the GUCY2D gene (NG_009092.1) used in Sanger sequencing were designed by Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/ primer-blast/) and synthesized by Sangon Biotech (Shanghai, China) (Table 1). All amplifications were examined by electrophoresis using 2% agarose gels and sequenced by BioSune Biotechnology Co., Ltd. (Shanghai, China). The sequencing results were further compared and analyzed by Mutation Surveyor [9].
Construction of wild-type (wt) and mutant human ROS-GC1 recombinant plasmids
The cDNA of human ROS-GC1 was obtained from Gene Copoeia (EX-Z0715-M98). Primers F and R containing XhoI and AgeI restriction sites were used to amplify ROS-GC1 (Table 2). The pEGFP-N1 vector was digested with XhoI and AgeI restriction enzymes. PCR amplification product was sub-cloned into pEGFP-N1 using ClonExpress (Vazyme, Nanjing, China) to give the recombinant plasmid pEGFP-GC1.
Site-directed mutagenesis PCR was used to construct the ROS-GC1 mutants. Primers used in site-directed mutagenesis PCR are shown in Table 2. Each mutant was achieved by two-step PCRs using pEGFP-GC1 as the template. For c.835G>A (Asp279Asn), two primer pairs F and r-835 and R-BamHI and f-835, were used in the first PCR step. Primers F and R-BamHI were used in the second step. For c.2783G>A (Gly928Glu), primers F-BamHI and r-2783 and R and f-2783 were used in the first PCR step. Primers F-BamHI and R were used in the second step. For each mutation, amplification products in the first step were cleaned (Axygen, CA, USA), mixed and used as the template in the second PCR reaction. All final PCR amplifications were ligated into the digested pEGFP-N1 vector using ClonExpress.
Recombinant plasmids pEGFP-GC1, pEGFP-Asp279Asn and pEGFP-Gly928Glu were transformed into Escherichia coli DH5a cells. DNA was prepared by using a plasmid DNA purification kit from Macherey-Nagel following the manufacturer’s instructions. Sanger sequencing was used to verify sequences.
Cellular localization of wt and mutant ROS-GC1 recombinant plasmids
pEGFP-N1, pEGFP-GC1, pEGFP-Asp279Asn and pEGFP-Gly928Glu were transfected into HeLa cells using the PolyJet Reagent (SigmaGen, MD, USA). After 36 h, cells were washed and fixed in 4% paraformaldehyde. The antibody anti-Na+/K+-ATPase (1:100, HuaAn Biotechnology Co., Ltd., Hangzhou, China) was used to identify the plasma membrane of HeLa cells. Details about the process have been described previously [10]. In HeLa cells, wt and mutant ROS-GC1 were co-express with EGFP, and the localization of pEGFP-N1, pEGFP-GC1, pEGFP-Asp279Asn and pEGFP-Gly928Glu was acquired by observing EGFP at 488nm excitation wavelength using a Nikon A1R.
Western blotting
Proteins from transfected HeLa cells were electrophoresed on a 12% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, MA, USA), and then incubated with the primary antibodies, anti-GFP (1:1000, Proteintech, Wuhan, China) or anti-glyceraldehyde 3 phosphate dehydrogenase (GAPDH) (1:5000, Abcam, Cambridge, UK) overnight at 4 °C. The PVDF membranes were washed with PBS and incubated with fluorescent secondary antibodies (1:1000, Abbkine, Wuhan, China) for 2 h at room temperature. The protein bands were visualized using an Odyssey Imager (Li-Cor Biosciences, NE, USA). GAPDH was used as the standard.
Validation of cGMP quantitation by HPLC-MS/MS
HeLa cells were transfected with pEGFP-N1, pEGFP-GC1, pEGFP-Asp279Asn and pEGFP-Gly928Glu. After 36 h, cells were collected from 100 mm plates and washed three times with PBS. The supernatant was removed carefully and 300 mL of ice-cold extraction medium (acetonitrile/methanol/water, 2/2/1 v/v/v) was added to each tube. Twenty-five ng/mL Tenofovir (TNF) was added as the internal standard. After dissolving, the sample was frozen immediately in liquid nitrogen for 30 s to terminate cGMP metabolism, and this was followed by incubation in a 37 °C water bath for 60 s. After repeating 6 times, samples were heated at 98 °C for 20 min. Samples were cooled on ice and centrifuged at 20,000 × g at 4 °C for 10 min. The supernatant was transferred into a new tube and the non-dissolved residue was extracted two more times with 400-μL extraction medium. After evaporation, residues were collected and dissolved in water for further analysis.
The cGMP concentrations were analyzed via HPLC-MS/MS. The samples were applied to an HPLC utilizing an ACQUITY CSH-C18 column (1.7 μm, 2.1 × 100 mm column, Waters, Ireland). The binary pump system supplied two eluents for chromatographic analysis, eluent A (10 mM formic acid) and eluent B (acetonitrile). The flow rate was 0.3 mL/min. Analyte detection was conducted on a sensitive triple quadrupole mass spectrometer (Waters TQ-XS, USA). Nitrogen was used as the collision gas. One-hundred ng/mL cGMP (G7504, Sigma, Germany) was used as the standard. All processes were referenced to the method described previously [11]. cGMP concentrations presented as means ± SEM are based on three independent experiments. P-values were calculated by means of the independent sample T-test.
Bioinformatics analysis
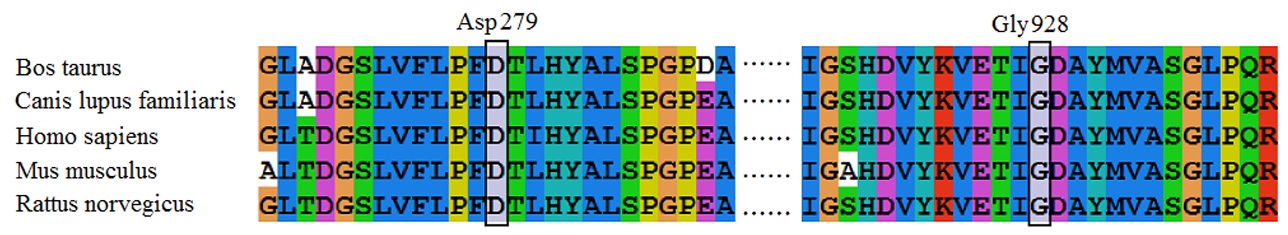
All sequences were analyzed by Mutation Surveyor software and aligned to the NCBI nucleotide sequence of GUCY2D (NG_009092.1). The pathogenicity of mutations was evaluated using the in silico predictors SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and Mutation Taster (http://www.mutationtaster.org/). Computational modeling of the mutant ROS-GC1 by Chimera (PDB ID: 1AWL) was carried out to study the effect of the Gly928Glu mutation on the three-dimensional (3D) structure of ROS-GC1.
{kind=link}
{kind=link}