Sample Characteristics
The clinicopathologic characteristics of the samples selected for this study are shown in Table 1. Among the full BBD Cohort, a frequency-matched case-control sample set of BBD biopsies was selected based on outcome in follow-up at 16 years: incident ER+ BC (BBD-ER+), incident ER- BC (BBD-ER-) or cancer-free (BBD-control), matched on age at biopsy and year of biopsy or censoring. Selection criteria also included availability of blocks with adequate tissue for DNA extraction. Severity of BBD was the only feature that differed significantly among the three groups (p=0.026). The BBD-controls included the highest percentage of non-proliferative disease (n=25; 59.5%), while the BBD-ER- group had the highest proportion of proliferative disease without atypia (n=19; 52.8%) and the BBD-ER+ group had the highest proportion of atypical hyperplasia (n=9; 21.4%), consistent with previous studies [5].
Gene-level associations
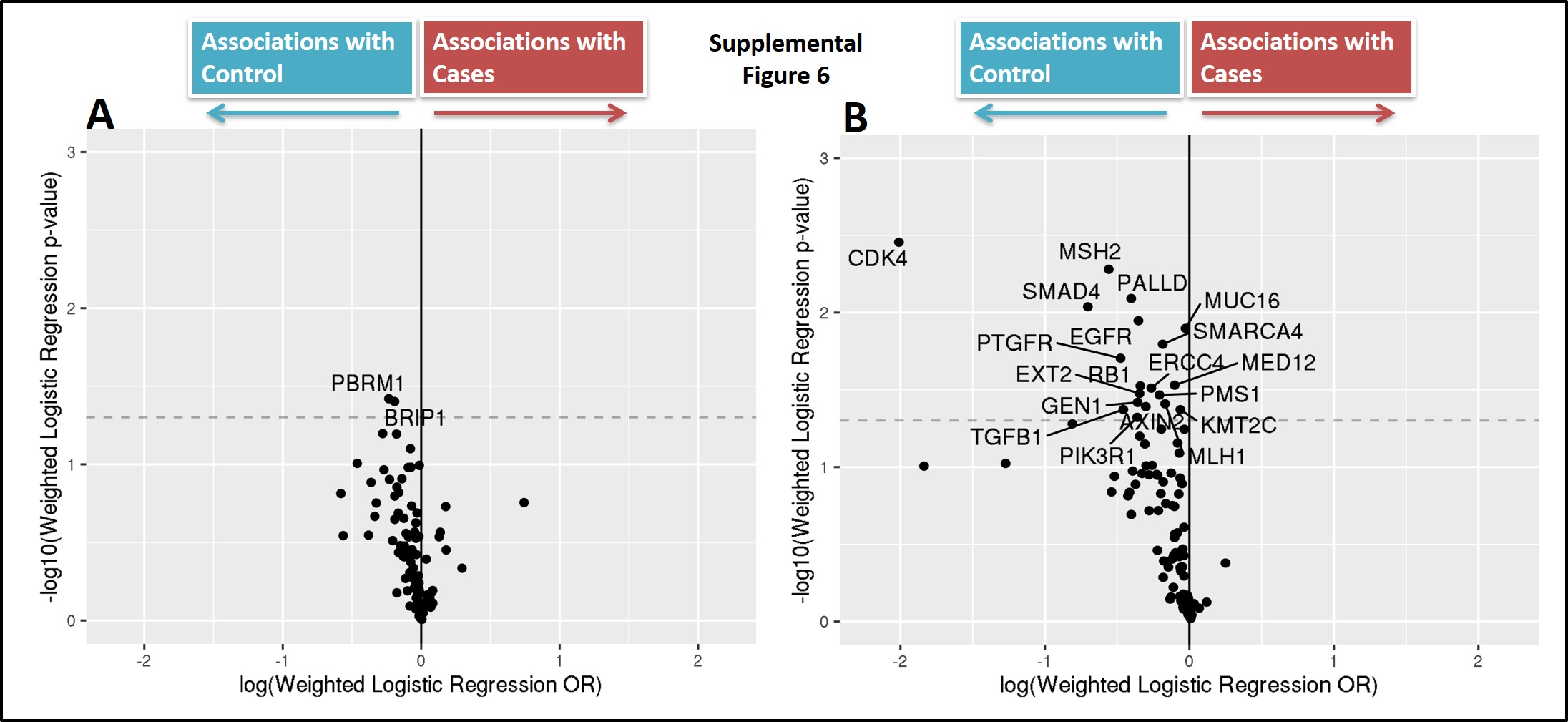
To address potentially artefactual FFPE variants, 12 combinations of variant filtering strategies and statistical analysis methods (classical, liberal and strict variant quality control (QC) filtering, combined with C-T weighted and un-weighted SKAT-O and logistic regression, described in Methods) were used to identify significant gene-level mutation burden differences between cancer (BBD-ER+ and BBD-ER-) and cancer-free (BBD-control) groups (Fig. 1A; Table 2). Full results are shown in Additional files 7-9. Through consensus analysis of association results shown as Fig. 1B, 10 genes (MED12, MSH2, BRIP1, PMS1, GATA3, MUC16, FAM175A, EXT2, MLH1 and TGFB1) of nominal significance (p<0.05) were found according to at least four methods.
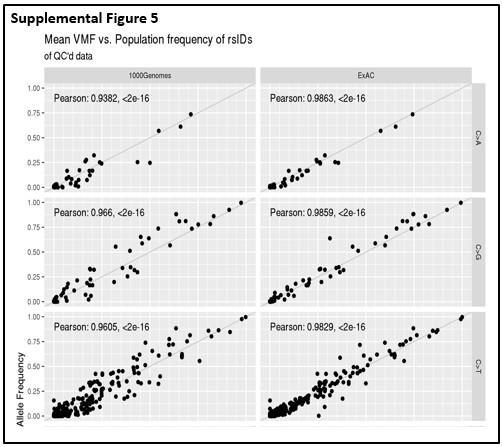
After extensive sample- and variant-level quality control, common single nucleotide polymorphisms (SNP) variants detected in this cohort had highly consistent allele frequency distributions when compared with population frequencies derived from large-scale germline sequencing studies such as 1000 Genome Project and Exome Aggregation Consortium (ExAC), shown as Fig. 2A. The concordance of detected allele frequencies with population frequencies were persistent even when variants were stratified by nucleotide substitution type [See Additional File 10]. This strong concordance at the population-level suggests a solid basis for the association analyses. When comparing overall BBD cancer cases (BBD-ER+ and BBD-ER-) versus BBD-controls, a volcano plot of gene-level association effect-sizes by the corresponding significance levels showed a skewed distribution, with more significant findings enriched for more mutations in cancer-free subjects (Fig. 2B). By further stratifying association analysis by type of BC (i.e. ER+ and ER-), the association differences were more profound when comparing BBD controls with BBD subjects with future ER- cancers than those with ER+ cancers, while the volcano plots remained skewed towards enrichment of protective associations [See Additional File 11].
Mutational Signatures
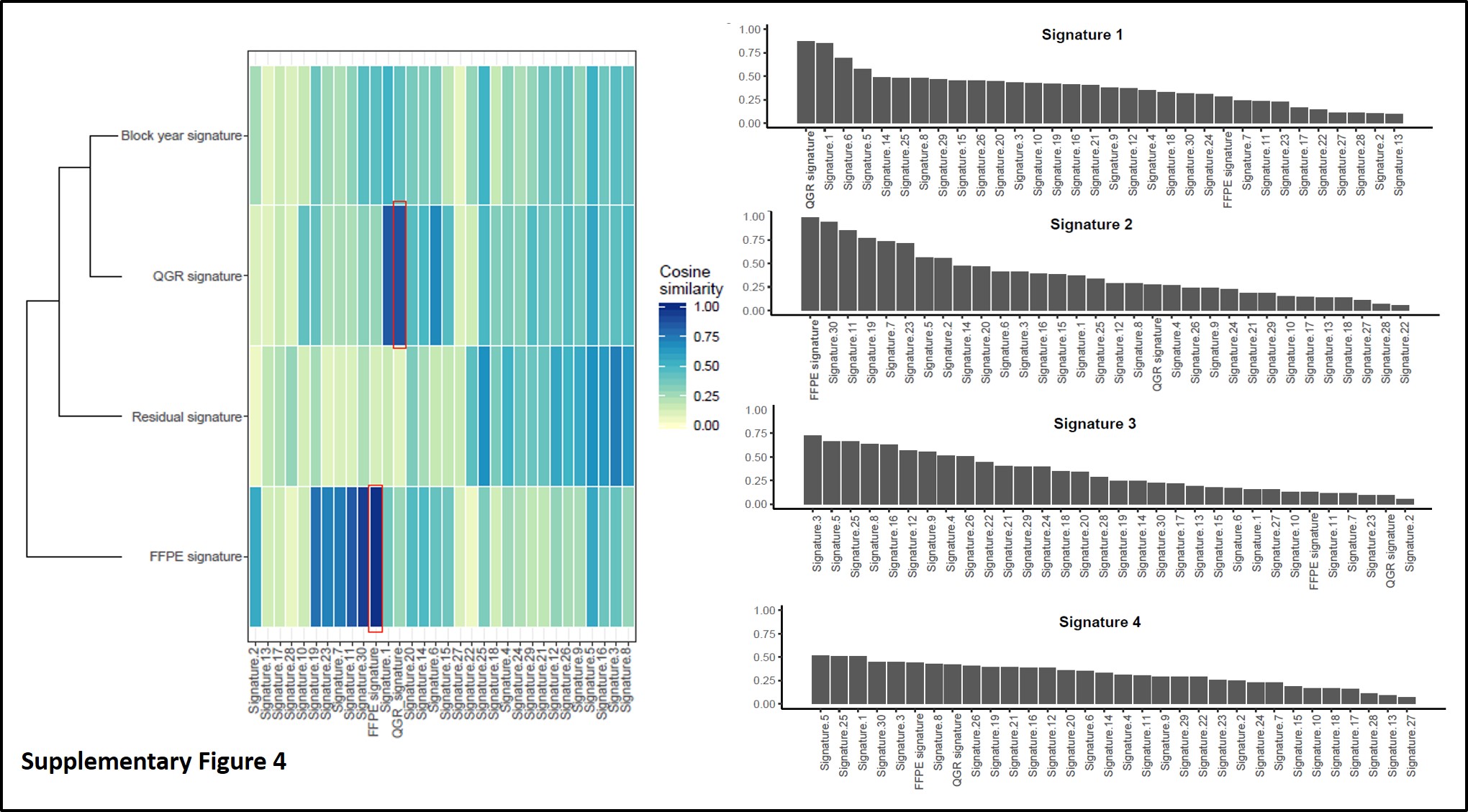
As we and others have shown, FFPE-derived sequencing may have distinct variant signatures collectively [15], and therefore de-novo mutational signature decomposition was conducted based on filtered variants for the entire BBD cohort, leading to four different mutational signatures shown in Fig. 3A: two of the observed signatures were primarily enriched for “C>T” paraffin artifacts and highly similar to FFPE/chemistry signatures, which we previously identified in paired comparisons between matched frozen and FFPE samples [15]. One of the de-novo signatures (Signature-D) was found to be highly correlated with collection age of FFPE block (Fig. 3B and 3C). However, no statistically significant difference was found between this block-year associated signature with cancer status (Fig. 3D). Nonetheless, this highlights the necessity of strict global variant quality control measures beyond variant-level checks for FFPE sequencing data. Furthermore, we assessed a previously published BBD signature that was associated with risk of triple negative BC for association in our dataset [13]; we did not observe presence of the signature in our sample, overall, or within ER+, ER-, or triple negative cases (p>0.05, data not shown).
Immunohistochemistry analysis
To follow-up on the findings that overall mutation was higher among BBD patients who remained cancer-free, we sought to investigate a potential hypothesis, where reduced mutational diversity is associated with 1) increased proliferation, or 2) reduced immune response. To investigate these hypotheses, we performed immunohistochemistry (IHC) analysis of Ki67 (as a marker of proliferation) and CD45 (as a marker of immune response) in normal lobules. Ki67 expression in normal lobules was very low, so analyses were not pursued further. However, CD45 expression was lower in BBD cases as compared to controls (p=0.19), although not statistically significant, and was positively associated with mutational burden (r=0.42, p=0.0031), most strongly in controls (r=0.50, p=0.005; Fig. 4 A-D).
Germline mutation information
Germline DNA was not available for the vast majority of subjects; however, a subset of 14 patients did have prior germline sequencing data available from other sources. Among these 14, 12 had no pathogenic mutations in pre-disposition genes. One BBD-ER- subject had a pathogenic mutation in BRCA1 that was also verified with clinical germline testing, and that BRCA1 mutation was also detected in the BBD tissue in this study. One BBD-ER+ subject had a pathogenic mutation in BLM but did not undergo confirmatory clinical germline testing, and the BLM mutation was not detected in the subject’s BBD tissue in this study.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}