Study design
This protocol will be designed as a randomized, placebo-controlled and multicenter trial. Participants, investigators and the statisticians will be blinded. 632 subjects will be recruited at the following 13 tertiary A hospitals in mainland China:Guang' anmen Hospital of the China Academy of Chinese Medical Sciences, The First Affiliated Hospital of Anhui University of Traditional Chinese Medicine, Beijing Hospital of Traditional Chinese Medicine, Hubei Hospital of Traditional Chinese Medicine, Zouping County Hospital of Traditional Chinese Medicine, Zibo Wanjie Hospital and Shijiazhuang Hospital of Traditional Chinese Medicine, Zhengzhou city Hospital of Traditional medicine, Xingtai city Hospital of Traditional medicine, Shexian country Hospital of Traditional medicine, Baoding city of Hosptial of Traditional medicine, Yantai bai shi Hospital of Traditional medicine, Jilin province of Hosptial of Traditional medicine. The trial will be implemented base on the principles of good clinical practice and reported according to the CONSORT statement [30,31]. The trial flow is illustrated in Fig. 1. The Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) [32] Checklist will be showed in Additional file 1. This study has been registered in ClinicalTrials.gov (NCT03009864).
Participants
Diagnostic criteria
The diagnostic criteria of this trial will be set based on the Chinese Diabetes Society guidelines [6] and American Diabetes Association guidelines [7],National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF-KDOQI) guidelines [33], and the Kidney Disease Improving Global Outcomes (KDIGO) 2012 Clinical Practice Guideline [34], For a diagnosis of early stage of DKD, patients with DM will have to have the following conditions:
- Diagnosed as type 2 diabetes.
- Type 2 diabetic patients with microalbuminuria and Urinary microalbumin excretion rate (UAER)
- For microalbuminuria, two repeated tests performed within six months will need to produce abnormal results;
- UAER of 30-300mg/24h or urinary albumin-to-creatinine ratio (ACR) of 30-300 mg/g (mg/mmol)
- Have clinical manifestations of kidney diseases, such as oedema, anaemia, renal dysfunction, etc.
Inclusion criteria
- The patient was diagnosed with DKD with microalbuminuria.
- Age in 30-70;
- Sign informed consent;
Exclusion criteria
- Proteinuria caused by non-diabetic kidney in the patient, such as gout, primary hypertension, tumors, and proteinuria caused by chronic kidney disease.
- Cardiovascular, liver, kidney, hematopoietic system, or other primary severe disease; serum transaminase more than double the standard value; serum creatinine (SCr) higher than the upper limit of normal; and psychiatric patients.
- Pregnancy, preparation for pregnancy, or lactation, or any history of drug allergy.
- The patient developed renal failure (anaemia and uremia).
- Participation in another clinical trial or use of any other drug within the previous month.
- Recent use of ACEI or ARB in the past one month except Losartan
- Any excessive consumption of alcohol or any consumption of psychoactive substances, drug
abuse, or drug dependence during the past five years.
- According to the researcher's judgment, some other diseases or conditions reduce the possibility of enrollment or complicate the enrollment, such as frequent changes of jobs and unstable living environment, which may easily lead to loss of visits.
Randomization and Concealment
Specific randomization sequence will be computer-generated by a computerized random number generator from an independent clinical research organization (CRO) of Institute of Clinical Medicine of Chinese Academy of Chinese Medical Sciences which is not involved in the study. All eligible patients will be randomized to the experimental group or the control group at a 1:1 ratio. An independent non-investigator will protect concealment list. The medical information will be confidential and not be available to any investigator for the duration of the study. If a medical emergency occurs, the individual's randomization code and group allocation can be identified.
Blinding
This study will be designed as a double-blind. Not only were subjects and investigators blinded in these trials, but drug administration, statisticians and curative evaluators were also masked. Treatment allocation will be uncovered after the completion of the study. In addition, TSF and placebo cannot be distinguished in the taste, smell, and appearance. After production, study drugs will be packaged and transferred to numbered bottles by designated pharmacists in accordance with the randomized list. No one can tell the difference except one who is in charge of concealment.
Intervention
All subjects in two groups will be received the conventional treatment, including oral Losartan (50mg/once a day), diet, exercise, and oral medicine, to ensure access to steady levels of blood glucose, blood lipids, and blood pressure and based on the recommendations of Treatment of Chinese Diabetes Society guidelines [6],and American Diabetes Association guidelines[7]. Subjects will be randomly assigned to receive placebo (6 g/bag twice per day) or Tangshan Formula (6 g/bag two times per day) by Specified sequence number from the central randomization system. The treatment will last for 24 consecutive weeks.
Outcomes
Primary outcomes
Urinary microalbumin (MAlb) is a critical indicator for the diagnosis of early renal impairment in diabetes mellitus. For early-stage DKD, Urinary microalbumin creatinine ratio (ACR) will be a primary evaluation index. The change in ACR from baseline will be evaluated between the two groups, and ACR will be compared at baseline and treatment endpoint (24 weeks) in each group.
Secondary outcomes
- Compare the ratio of progression into the clinical proteinuria period after intervention between the two groups
- Urine microalbumin negative conversion rate. The ratio of normal urinary microalbumin (<20 ug/min) was compared between the two groups.
- Change in GFR. Change in the D-value and ratio of glomerular filtration rate (GFR) before and after treatment were compared between the two groups. Glomerular rate filtration (GFR): using the simplified MDRD formula: GFR (ml/min·1.73m2) =186×serum creatinine-1.154×age-1.154× [female×0.742] × [Chinese×1.233].
- Doubling rate of baseline creatinine value will be compared between the two groups after intervention
Safety assessment
Adverse events(AEs)will be continuously monitored for 24 weeks at the beginning and end of the study period, and the incidence of AEs will be evaluated in each visit, including vital signs, ECG, liver function, renal function, Routine blood examination, routine urinalysis, and routine stool examination. In addition, AEs, such as signs and symptoms and other ailments, will also be documented truthfully at every study visit, including the occurrence time, severity, duration, effective measures and transfer. Each AE associated with the intervention drugs will be classified as mild, moderate and severe. Severe AEs will need to submit to the principal investigator and the ethics committee within 24 h. All AEs will be properly resolved. Criteria for the severity of adverse events, detailed requirements are as follows:
Mild: mild discomfort, subjects can endure no treatment, no special treatment is required, no effect on subjects' recovery.
Moderate: moderate discomfort, unbearable to the subject, requiring special treatment, has a direct impact on the subject's recovery.
Severity: severe discomfort, life-threatening, fatal or disabling, immediate emergency treatment is required.
Study visits and assessment
Intervention cycle will be 24 weeks, including a run-in period. After the research commences, visits will occur every four weeks during the study period. An overview of specific measurements and visit of time points for data collection can be shown in Table.1.
Quality control data collection
To maintain the high quality of this trial and assure its adherence to the protocol, all investigators and drug administrators participating in the research will be trained rigorously basis on a standardized operation practice (SOP) manual. Withdrawals or drop visits also will need to be explained in CRFs. All data will be documented in a standardized case report form (CRF) and instantly recorded in the database via the ClinResearch Electronic Data Capture System at http://www.tcmcec.net/crivrs/. The monitor will review the CRFs, check the inclusion, exclusion, and withdraw criteria, as well as ensure information of CRFs in accordance with those in the source medical records. Except for, Original CRFs will be reserved at the research centre for five years after completion of the study. The validity and authenticity of the multicenter trial will be guaranteed by establishing three committees including the clinical trial guidance committee, the data and safety monitoring board and the outcome evaluation committee, hitch respectively being responsible for trial design and the executing process, monitoring the data collection process to control its quality and evaluating the outcomes. The diagram of enrollment, interventions, and assessments is shown in Fig.1.
Sample size
The sample size was estimated according to the relevant data from Cossar study published in New England [18,19]. These results showed that the proportion of patients with ACR reduced by 50% or more in the Cosszar group was 12.5% and the preliminary study of TSF data manifested that TSF as an add on study can improve 50% of patients. Estimating the proportion of patients with ACR reduced by 50% or more was 18.7%.The estimated sample size formula was tested by using the hypothesis of two population rates:(See Formula 1 in the Supplementary Files)
(n is the sample size; p1 and p2 are the sample rate, and p= (p1+p2) /2 is the sample average rate, α is the type 1 error and β are the type 2 error, while uα and uβ are the locus of the corresponding standard normal distribution). According to the unilateral test, uα=1.64485, uβ=0.84162, and substitute it into above formula, n=274.46. Therefore, 275 patients were needed in each group. Considering the drop-rate of no more than 15%, the final sample size was estimated to be 632 in total.
Statistical analysis
The statistical analysis of this study was completed by independent statistician, and the detailed statistical analysis plan was formulated separately by statistician before the inception of this trial and determined with the principal investigator. Three analysis sets will be used for assessment of this study: the intention-to-treat set (ITT), the per-protocol analysis set (PPS) and the safety analysis set (SAS). ITT and PPS will be used to appraise the efficacy of TSF. If any given case exists missing a critical variable, the last observation used as the final results will be carried forward to the finial data. The changes of urinary microalbumin creatinine ratio, urinary microalbumin, GFR, creatinine and baseline information will be present after treatment. A paired t -test or the Wilcoxon signed-rank test will be employed to compare each group. Changes relative to baseline after treatment will be compared between groups using the t-test or the Wilcoxon rank-sum test. A P value < 0.05 will be deem as to show a statistically significant difference.
Bias analysis
The main evaluation outcomes of this trial are the rate of progression from microalbuminuria period into clinical proteinuria, which are very objective. Although ACEI and ARB drugs have achieved a recognized efficacy internationally, a certain proportion of patients still cannot delay the progression of DKD even using these two drugs. Therefore, the bias factors affecting the outcome evaluation include three aspects:(1) blood glucose level (2) ACEI or/and ARB use and (3) Laboratory testing error of urine microalbuminuria.
These factors are solved as follows :(1) for the blood glucose level, the consistency of this factor in the two groups was ensured due to the random and double-blind study design method. (2) subjects only took cossar and no other ARB or/and ACEI drugs. (3) Central laboratories are used for the main outcomes.
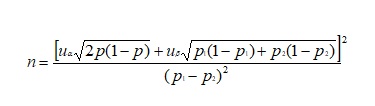{kind=link}