Transcriptome data assembly and acquisition
In general, the high-quality sequence (6.47 GB) comes from the transcriptome sequencing of T. hemprichii. The average error rate of the sequences was 0.01%, and more than 87% of clean reads (Figure 1A). Using Benchmarking Universal Single-Copy Orthologs (BUSCO) v3, a single copy homologous database, and the integrity of the transcriptome assembly was demonstrated by comparing with the conserved genes (Figure 1B). The assembled sequence data for these raw reads were deposited in the Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/Traces/sra) (Accession no. SAMN13255716 and SAMN13255717). The sequencing data were assembled into 51,619 transcripts with the sequencing lengths ranging from 200 to 19,192 bases (Figure 1C) (mean length = 1045 bases, N50 = 1798 bases, and GC content = 45.73%). And the longest Unigene is 5.40 Mb (53,976,312 bases). All the data show that the production capacity of the product is high enough and the assembly quality is very good, which can be used for subsequent analysis.
Gene annotation and functional classification
Seven databases (KEGG), Gene Oncology (GO), NR, NT, SwissProt(SWISS-PROT), Protein families (Pfam), and EuKaryotic Orthologous Groups (KOG) were used to annotate the obtained genes. In total, 32,097 Unigenes (62.18% of the total Unigenes) were annotated in the databases in this study (Table 1). Transcoder software was used to identify the candidate coding regions (CDs) in Unigene. First, the longest open reading frame was obtained. Then, through Swissport database and blast in hmmscan, the homologous sequence of Pfam protein database was found and its CDs was predicted.. 24,870 CDS were predicted, with a total length of 26,466,750, N50 of 1,407 bases, length range of 297 to 10,956 bases and GC content of 49.09% (Figure 1D). According to the results, 11,804 genes were annotated in all databases. Most of the unigenes get annotation information in the NR database (30,772 and 59.52%) (Figure 2). Regarding to GO classification, the highest number of annotations was in the molecular function (MF) category, for which catalytic activity (6317), binding (6269) and transporter activity (676) were the top 3 GO terms; the second-largest amount of annotations was in the cellular component (CC) category, the top 3 GO terms were membrane part (3948), cell (3746) and organelle part (1512); and the third-largest amount of annotations was in the biological process (BP) category, the top 3 GO terms were cellular process (3613), biological regulation (1404) and localization (879) (Figure 3A). According to the classification of KEGG, the most annotated genes are metabolic related pathways, and the most annotated pathway is metabolic pathway (15,152). In this category, the Global and overview maps pathway enriched the most genes (5,855) (Figure 3B). Furthermore, Classification of Unigene according to KOG. The top 3 classes were general function prediction(5,970), signal transduction mechanisms(3,334), and function unknown(2,071) (Figure 3C).
Functional homology analysis of whole transcriptome
In the results of gene function annotation, NR database had 59.52% of the annotation proportion. The proportion of different species on the NR annotation was calculated, and the species distribution map was drawn (Figure 4). The NR annotation of top 5 species were Elaeis guineensis (5,272 and 17.16%), Phoenix dactylifera (3,436 and 11.19%), Ananas comosus (1,896 and 6.17%), Nelumbo nucifera (1,756 and 5.72%) and Zostera marina (1,672 and 5.44%). T. hemprichii and E. guineensis had more closely related genes and were closer to the terrestrial plants.
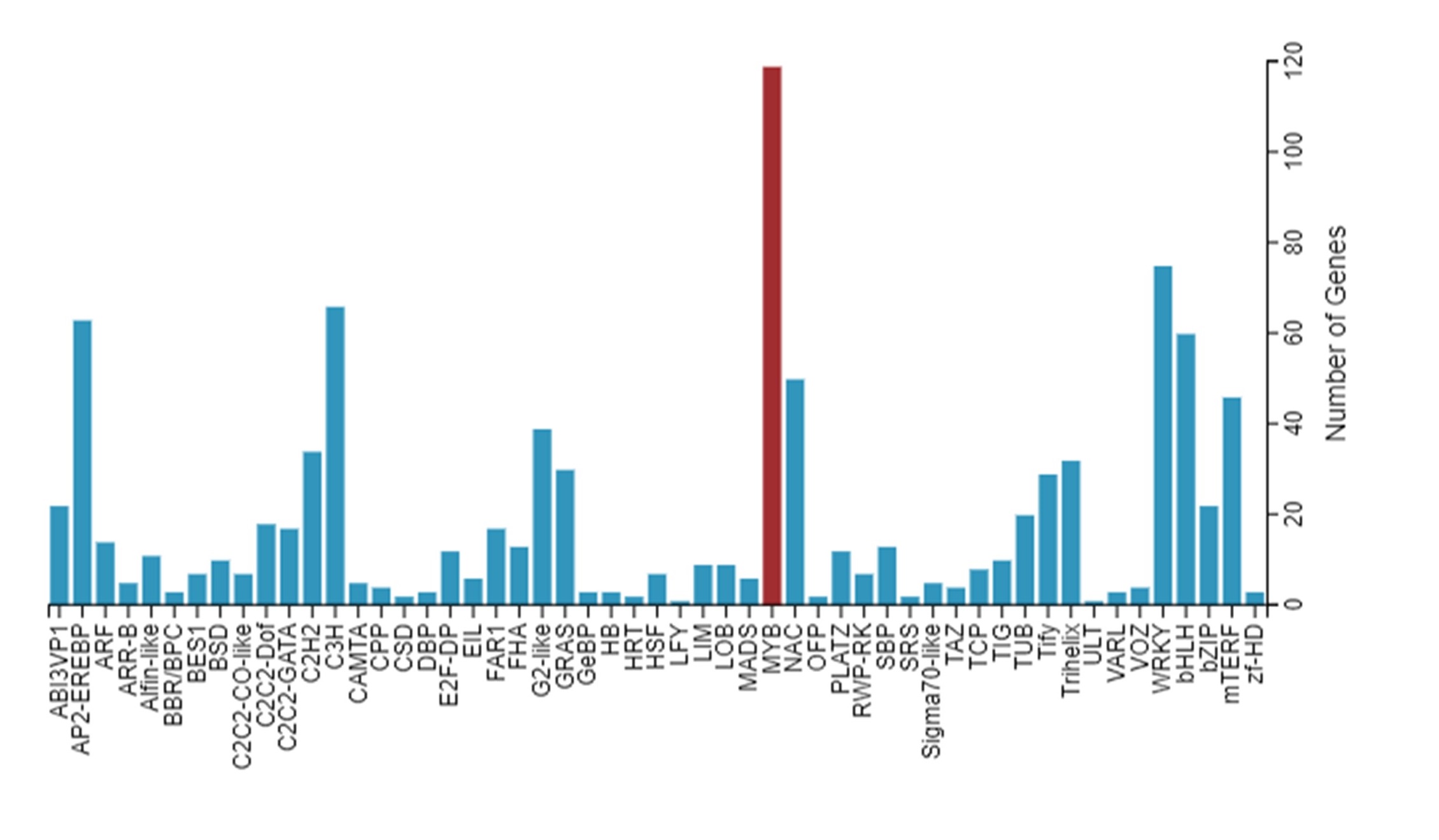
Identification and distribution of MYB TFs encoding genes in transcriptome
The genes were predicted, which were capable of encoding transcription factors (TFs). Then,we use getorf to detect the ORF of Unigene, and then useing hmmsearch to compare ORF with transcription factor protein domain (the data comes from TF), and then recognize the ability of UniGene according to the characteristics of transcription factor family described by TFDB. The TFs family was classified and counted (Additional files 1: Figure S1). There were 119 MYB genes family in all TF genes (Additional files 2: Table S1).
GO classification was used; the results showed that these MYB family genes were divided into 10 subclasses. The most annotated genes was in the molecular function (MF) category; binding (69), and transcription regulator activity (13). In the cellular component (CC) category, the 3 GO terms were cell (42), organelle part (2), and membrane part (1). In the biological process (BP) category, the 5 GO terms were cellular process (39), biological regulation (39), response to a stimulus (6), cellular component organization or biogenesis (2), and multicellular organismal process (2) (Figure 5A). According to the KEGG classification, All MYB family genes were involved in 9 pathways, among which these pathway was Environmental adaptation (24 genes), Global and overview maps (9 genes), Biosynthesis of other secondary metabolites (4 genes), Transport and catabolism (4 genes), Lipid metabolism (3 genes), Translation (1 gene), Transcription (1 gene), Carbohydrate metabolism (1 gene), and Energy metabolism (1 gene) (Figure 5B). Also, 24 of these genes were enriched in the Circuit rhythm pathway.
conserved DNA-binding domain analysis and of Protein characteristics MYB TFs.
Through comparative analysis, a set of 119 MYB Unigenes containing MYB DNA-binding domains was identified in T. hemprichii (Additional files 3: TableS 2), which included 67 full-length CDS and 52 fragment sequences. MYB Unigenes (ThMYB1–ThMYB119) were identified, respectively. The full-length sequence of MYB ranges from 588 to 3393. The amino acid sequence length of the protein is 148-1131 amino acids, the calculated molecular weight of ThMYB is 15.87-127.50 kDa, and the calculated pi is 4.34-110.01. The majority of ThMYB protein is about 400 amino acids, and its molecular weight is about 50 kDa. Among the 119 ThMYBs, ThMYB042 was the longest protein (1,131 amino acids), while the shortest protein was ThMYB038 (148 amino acids).
In order to study and identify the characteristics of homologous domain of ThMYB protein, 119 amino acid sequences of ThMYBs were used for multiple sequence alignment. According to the domain classification, ThMYB protein contains type 11 domain, which shows that ThMYBs has similar domain with other species (Figure 6).
Putative functions of ThMYBs in T. hemprichii
MYB TFs in A. thaliana contains 27 clades, and the function of each clades was annotated [14, 18-21]. The conglomerated homologous proteins usually have similar functions, which indicates that ThMYB has similar functions with AtMYB in the same clades. Therefore, through the conclusion and discussion compared with AtMYBs, the function of ThMYBs is predicted and summarized (Figure 7; Additional files 4: Table S3). Constructing NJ unrooted phylogenetic tree with 09 ThMYBs and 110 AtMYBs (Figure 7). The results showed that MYB TFs of the two species gathered in 36 clades (C1-C36), and 17 of which were found in both species.
{kind=link}