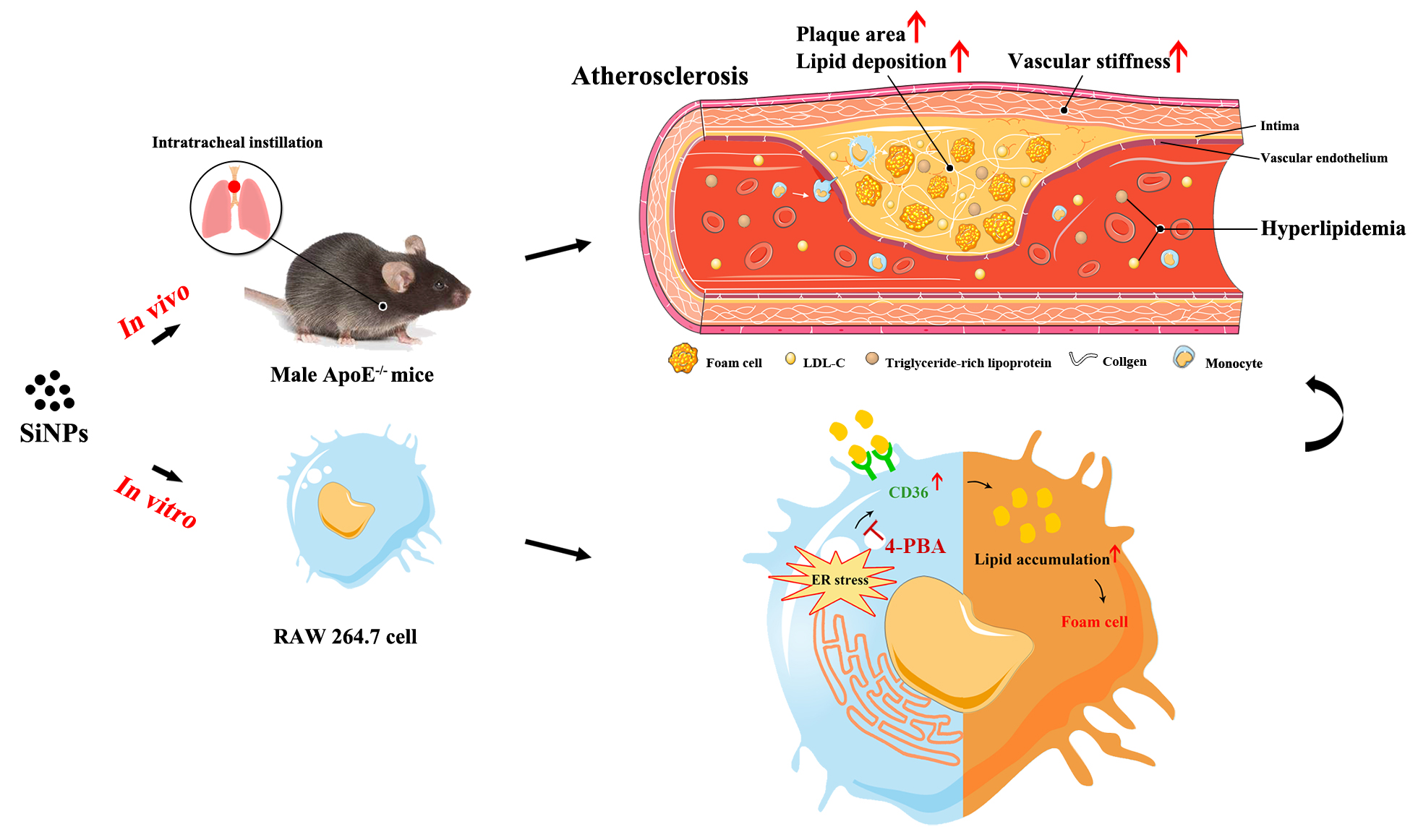
Atherosclerotic related cardiovascular disease is the leading cause of mortality worldwide. So far, there is still no conclusive information on the pro-atherogenic potential of SiNPs. To the best of our knowledge, this is the first in vivo study to confirm SiNPs exposure could indeed accelerate the progression of atherosclerosis. We firstly administered Western diet-fed ApoE−/− mice with SiNPs or saline through intratracheal instillation for 12 weeks, and ultrasound biomicroscopy (UBM) was applied to monitor the development and progression of atherosclerosis. Epidemiological studies have provided a close correlation between PM2.5 and carotid intima-media thickness (CIMT), a most frequently used indicator of subclinical atherosclerosis [26]. However, only few studies reported the vascular effects of NPs through Doppler ultrasound trace. Here, ultrasound technique was applied, and repeated measurement data was acquired, which would be more convincing. As a result, the data indicated the atherosclerosis model was well established, and SiNPs exposure reduced arterial elasticity, aggravated the arterial stiffness, which is a feature of atherosclerotic plaque progression [27]. Arterial stiffness was positively correlated with reactive oxygen species (ROS) and the followed oxidative stress [28], a major attributor of the adverse effects caused by SiNPs, and inversely with arterial NO bioavailability. A declined NO bioavailability was detected as a decrease in endothelium-dependent vasodilatation [29]. Indeed, a disturbed oxidation-reduction status was triggered after intratreacheally instilled SiNPs [21], as well as NO/NOS disorder.
On the basis of a clearly observed plaque and an increased PWV value in SiNPs-treated mice, the experiment was terminated, and atherosclerotic burden was assessed by en-face staining of the whole aorta and histological examination of the aortic root. Consistent with the UBM monitoring, plaque was formed in aorta, and the plaque area and lipid-rich core area in plaque were increased after SiNPs administration. Plaques with lipid-rich cores are suspected to be vulnerable, increasing the risk of sudden plaque rupture and thrombotic complications. Previous studies have found the induction of systemic inflammation by SiNPs [30], probably contributing to the form of vulnerable plaques [31]. In addition, according to the American Heart Association's definition of human atherosclerotic stages [32, 33], the plaque at the termination of experiment was progressed into advanced lesion with stage IV, characterized with the large plaque burden and foam cells, migrating smooth muscle cells, cholesterol crystals, and necrotic substances inside the plaque. Previously, SiNPs was confirmed to induce endothelial injury [34, 35, 36, 37] and promote the recruitment of monocytes to injured endothelial cells [38], and foam cell formation at the early stage of atherosclerosis [39]. Moreover, a discontinued or fragmented intimal surface was induced by repeated pulmonary exposure of SiNPs, accompanied with endothelial apoptosis [21]. In addition, the ability to induce thrombosis formation may also attribute to the pro-atherogenic potential of SiNPs [40]. However, the underlying mechanisms by which SiNPs influenced atherogenesis still remains largely unknown.
Lipid is one of the most important stimuli initiating atherogenesis. Plasma lipoproteins are involved in foam cell formation and inflammatory regulation within plaques [41, 42, 43]. The abnormal plasma lipoprotein level is usually considered as the feature of atherosclerosis. To date, few studies have pointed out the induction of dyslipidemia by airborne UFP [44]. Repeated intravenous administration of SiNPs was reported to disturb hepatic lipid metabolism and trigger hyperlipidemia in mice [45]. Similarly, dys- or hyper-lipidemia was induced after a long-term exposure of ZnO NPs via intratracheally instillation in rat, or of TiO2 NPs in ICR or ApoE−/− mice, ultimately contributing to the initiation and progression of atherosclerosis [46, 47, 48, 49]. In particular, a cross-sectional study also found that occupational exposure to TiO2 NPs affected lipid metabolism, as evidenced by a higher level of serum LDL when compared to the normal physiologic range for LDL in adults in China [50]. In agreement with these findings, we revealed the sub-chronic SiNPs treatment via intratracheal instillation also aggravated the hyperlipidemia of ApoE−/− mice, characterized with the increased serum levels of TG and LDL-C. To be noted, the increase in serum LDL-C content was positively correlated with the plaque area in aortic root. Coincidently, population studies have shown a highly consistent, positive correlation between blood LDL-C level and atherosclerotic CVD risk in humans [51]. Besides, hypertriglyceridemia is causally associated with increased atherosclerosis risk [42]. Intriguingly, either the repeated exposure to single-wall carbon nanotube (SWCNT) or long-term (over 5 months) exposure of nano-Ni was reported to exacerbate plaque development in ApoE−/− mice, but no alteration of lipid profile [49, 52]. It might explain other mechanisms also contributing to atherogenesis, such as systemic oxidative stress, inflammation [53].
Endoplasmic reticulum (ER) stress, also known as unfold protein response (UPR), plays a crucial role in the pathogenesis of a series of cardiovascular disorders, including atherosclerosis, ischemia. ER is a major site for protein folding and calcium reservoir. Numerous studies suggested ER as a potential target for NPs [34, 54, 55, 56, 57, 58]. The accumulation of misfolded or unfolded proteins led to ER stress, which was recently proposed as the mechanism responsible for NPs-induced toxicity [59]. ER stress could occur at all stages of atherogenesis [60]. Moreover, studies by using human plaque samples and animal models showed an increased ER stress in plaques with faster plaque progression [61]. The induction of ER-associated UPR events by NPs was pointed out either in vitro or in vivo [34, 62]. In particular, other than oxidative stress, ER stress was reported to mediate the SiNPs-caused vascular injury in rats [21], and modulate ROS formation in vascular endothelial cells, which could be explained by the increased ROS as an integral component of UPR signaling [63]. At the present study, aggravated ER stress in macrophage within plaque was clearly seen, as evidenced by ER expansion and remarkably elevated Bip and CHOP levels. But how ER stress participates and its regulatory molecular mechanism among the pro-atherogenesis effect of SiNPs was still unclearly illuminated.
Macrophage-derived foam cell is the crucial determinant of the initiation and progression of atherosclerosis lesion [64], contributing to plaque instability and rupture [65]. In agreement with previous studies [39], ER stress was significantly induced after SiNPs exposure, as evidenced by the expansion and degranulation of ER, as well as greatly up-regulated Bip and CHOP expressions. More importantly, ER stress inhibition largely alleviated the lipid accumulation induced by SiNPs in macrophage as assessed by the Oil-Red O staining and intracellular cholesterol measurement. It is worth noting that some ER stress genes are involved in lipid metabolism, which fuel atherogenesis. For instance, CHOP is crucial for lipid synthesis, and its expression would result in lipid accumulation [66]. The cellular lipid homeostasis was highly, precisely regulated by lipid influx and efflux. Upon oxLDL and SiNPs co-exposure, remarkably dysregulated expressions of lipid influx/efflux genes were detected in macrophage [39], probably mediated by ER stress signaling. Further, Long et al. revealed the lipid accumulation in macrophages was attributed to the modulation of ER stress leading to the upregulation of scavenger receptors, including CD36 and SRA1 [67]. Apart from CD36, ER stress was also reported to correlate with reduced ABCA1 level in macrophage, a key regulator for lipid efflux. But that was not the case in SiNPs-induced lipid accumulation. However, our data confirmed only CD36 was dependent on ER stress induced by SiNPs, leading to lipid accumulation and foam cell formation, ultimately contributing to atherosclerosis. CD36 is a membrane glycoprotein that belongs to the class B scavenger receptor family, and is known to be involved in lipid metabolism as well as atherosclerosis development. Indeed, a series of studies have proven ER stress modulated lipid influx via CD36 [70, 71, 72]. Lipid could be trafficked by CD36 to ER, and meanwhile, the accumulation of toxic lipids in macrophages would result in a prolonged ER stress. However, the detailed molecular mechanisms by which ER stress regulate CD36 need to address in future studies. Of note, the enhanced intracellular cholesterol content and the modulation of cholesterol influx/efflux genes could be directly induced by SiNPs even without external lipid supply, indicating the excessive lipids are not indispensable for SiNPs to promote foam cell formation.
By the way, how do the applied doses relate to a real-life exposure? The air concentrations of amorphous SiNPs ranged 400–22200 particles/cm3 [73]. The number of NPs in ambient air ranged from 2 × 104 to 2 × 105/cm3, with mass concentrations of > 50 µg/m3 near major highways [74]. Here, the actual lung exposure dose of SiNPs (1.5, 3.0 or 6.0 mg/kg·bw) was about 40, 80, 160 µg/mouse/week (based on mice weighting 26–29 µg during SiNPs treatment), respectively. According to a previous study [48], the inhalation dose of a mouse is about 5 µg after a one-week exposure at the daily concentration of 50 µg/m3 near major highways (consuming the inhalation rate for mice is 0.052 m3/day, and the mice pulmonary deposition fraction for 60 nm particles of 0.25) [75]. In this concentration, the applied dose (1.5, 3.0 or 6.0 mg/kg·bw) in this study was correspondingly 8, 16, or 32 times to the airborne exposure level of SiNPs. Hence, this study is featured by the used exposure conditions relevant to human exposure scenarios.

{kind=link}