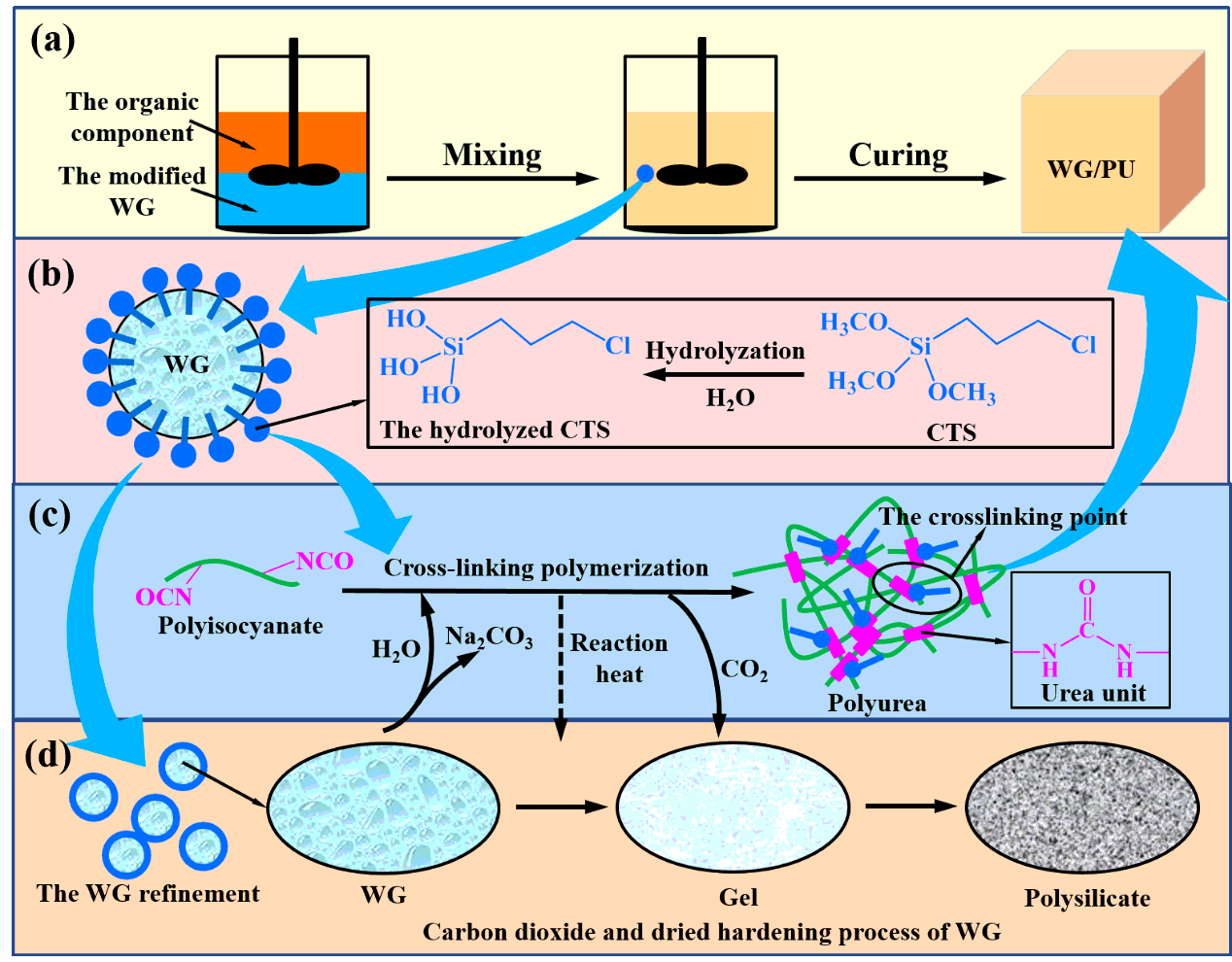
3.1 Synthesis mechanism of CTS modified WG/PU composites
Scheme 1 schematically shows the primary chemical reactions route that occurs during the WG/PU composites preparation and the CTS effect. The organic component and the modified WG are mixed using a two-blade paddle mixer. The achieved homogeneous mixed slurry is poured into the mold and curied until theexpected WG/PU composites is achieved (see Scheme 1a). During the process of mixing, the hydrolyzed CTS is oriented at the interface between the organic phase and the inorganic phase, thus forming a smaller and more uniformed inorganic dispersion system by the reduction of the organic-inorganic interfacial tension (see Scheme 1b and d). This benefits the acceleration of the curing process and the better dispersion of the stress. Thus, the mechanical properties of the composites are further improved. During the following curing of WG/PU composites, multifunctional polyisocyanate is reacted with the water from the WG, leading to a highly cross-linking network of formed polyurea by urea units in the organic phase (see Scheme 1c). More water consumption produces more carbon dioxide into WG, which in turn produces more sodium carbonate, leading to gelation of the WG in the inorganic phase. Water continuously enters the organic phase from the inorganic phase, while the diffusion path of the produced carbon dioxide is opposite. The directional mass transfer of water and carbon dioxide together realizes the hardening of the WG to the polysilicate and the cross-linking polymerization of the organic phase, and induces the solidification of of the organic phase and the inorganic phase eventually. Moreover, a large amount of reaction heat from the amine-isocyanate chemistry will also accelerate this curing process. In addition, the lipophilic group can make part of CTS get into the organic phase well-distributed. Subsequently, the reactive lipophilic groups of the CTS as hard segment will bind with the crosslinking point (e.g. the NCO and urea groups) of the organic phase, forming a chemical bond. Therefore, it is precisely because of multiple physical and chemical interactions between the CTS and the WG/PU composites that the WG/PU composites possess the outstanding mechanical properties.
3.2 Contact angle analysis of the modified WG
To achieve high mechanical performance of the composite, interface properties between the organic and the inorganic phases need to be improved. Figure 1 shows the static contact angle of the modified WG on the cured organic component surface. Pure WG presents a contact angle of 117.49º, whereas by modifying it is gradually reduced from 117.49º to 77.73º. An improvement of more than 33% is observed in wettability of WG on the cured organic component surface. It is probably ascribed to the presence of lipophilic CTS on the WG surface. This result is confirmed by modification, which makes WG more lipophilic. This improved wetting property is beneficial to the refinement and uniform distribution of the WG in PU matrix and improves the strength of the WG/PU composites.
3.3 Mechanical properties of WG/PU composites
Figure 2 shows that the compressive strength obtained for the WG/PU composites with the CTS content of 0.0 wt%, 0.5 wt%, 1.5 wt%, 2.5 wt% and 3.5 wt%. The CTS is observed to enhance the compressive strength of the WG/PU composites significantly. Excellent wettability allows maximum inorganic phase refinement. Accordingly, the curing process of the WG/PU composites becomes faster and more complete. Therefore, the compressive strength of the WG/PU composites varies from 55.07 MPa to 62.19 MPa with the increase of the CTS content from 0.0 wt% to 2.5 wt%. As CTS content is greater than 2.5 wt%, a peak is reached for the WG/PU composites, showing a slight decrease in compressive strength thereafter. This is probably due to the arising interface saturation with the CTS in the WG/PU composites. The outcome therefore indicates that the interface saturation between the inorganic phase and organic phase may start as early as the 2.5 wt% CTS content range, which is adequate to reach high compressive performance.
To further understand the mechanical properties of the WG/PU composites, the flexural strength, flexural modulus and fracture toughness of the WG/PU composites with 2.5% of CTS optimal addition and without CTS are examined respectively, as shown in Figure 3. From Figure 3, it is observed that 2.5 wt% of the CTS appropriately improves the average flexural strength of the WG/PU composites from 39.3 MPa to 41.9 MPa. However, the 17.5 % and 9.7% enhancement in flexural modulus and fracture toughness are achieved from 2.5 wt% CTS modified the WG/PU composites, respectively. This may be attributed to the active crosslinking effect of the CTS. The CTS can enter and form strong chemical interactions with the organic phase of the WG/PU composites and further strengthen the interior crosslinking structure of the organic phase, thus facilitating the improvement of strength and toughness for the WG/PU composites. Therefore, 2.5 wt% of CTS will be used as the designated amount of modified WG/PU composites. The obtained WG/PU composites will be used for the fracture surface morphology, elemental composition, chemical structure and thermal properties analysis.
3.4 SEM-EDS analysis of WG/PU composites
In order to understand constitutes of the WG/PU composites and further explore how the CTS induces physical and chemical interactions to enhance the mechanical performance of the WG/PU composites, SEM and EDS are used together to observe the fracture surface morphology and analyze the elemental composition and content in the WG/PU composites. The SEM images show the spherical dispersed phases embedded in the the continuous phase. Moreover, the continuous phases exhibit many dome-shaped cavities throughout the fractured surface of the compsites. It is obvious that there is insufficient interfacial bond strength between the continuous phase and the dispersed phase. The EDS analyses show that the continuous phases consist of C (65.54 wt%), O (11.38 wt%), Cl (13.17 wt%) and C (70.11 wt%), O (13.23 wt%), Cl (12.98 wt%) in the non-modified and modified WG/PU composites, respectively. It reveals the continuous phase originated from the organic component. On the other hand, the main elements of the dispersed phase are Si (32.15 wt%), O (44.96 wt%), Na (12.57 wt%), C (10.33 wt%) and Si (30.10 wt%), O (46.65 wt%), Na (10.04 wt%), C (13.21 wt%) in the non-modified and modified WG/PU composites, respectively. The results therefore prove that the dispersed phase originates from the WG.
As shown in Figure 4A(a) and B(a), compared with the non-modified samples, 2.17 wt% of the Si element appears in the continuous phase of the modified sample. This phenomenon illustrates that the CTS will migrate into the organic phase in the modified WG/PU composites. Figure 4A(b) and B(b) show that the element content of C (42.99 wt%), Cl (24.90 wt%) and Si (2.78 wt%) on the cavity surface of the modified composites are much higher than that (C (21.09 wt%), Cl (10.75 wt%) and Si (trace))of the non-modified composites. This confirms that the CTS is widely distributed at the interface between the organic phase and the inorganic phase. Furthermore, it is found that C and Cl elements are greatly increased on the cavity inner surface of the modified WG/PU composite. This demonstrates that the introduction of CTS improves the interface compatibility and affinity between organic and inorganic phases. In addition, it can be clearly seen that the O (46.65 wt%) and C (13.21 wt%) content on the dispersed phase of the modified sample are higher than that (O (44.96 wt%) and C (10.33 wt%)) of the non-modified sample (see Figure 5A(c) and B(c)). This increment is primarily attributable to higher CO2 accumulation rates at the inorganic-organic phase interface caused by the rapid cross-linking polymerization of the organic phase in the modified WG/PU composites.
In order to confirm the influence of CTS on the distribution and morphology of WG in the PU matrix. The cross-sectional SEM images of the non-modified and modified WG/PU composites are shown in Figure 5a and b. The SEM images confirm that the polysilicate particles are individually dispersed in the PU matrix. Moreover, the SEM images show that the polysilicate particle size and dome-shaped cavities have become smaller and more uniform in the modified WG/PU composites due to the addition of CTS, which is responsible for the relatively high mechanical strength and toughtness of WG/PU composites. In addition, Figure 5c and d directly show the size distribution histograms of the polysilicate particles of the non-modified and modified samples. The size of polysilicate particle varies from 0 μm to 60 μm, and 0 μm to 24 μm for the non-modified and modified samples, respectively. Moreover, the mean size of polysilicate particle for the non-modified and modified samples are also reduced from 11.3 μm to 5.7 μm. The results demonstrate that the CTS has excellent emulsification ability. Additionally, 4.4% of the polysilicate particles with a scale between 24 μm to 60 μm are present in non-modified samples. Increasing the size of polysilicate particles leads to larger cavities formation. These particles and cavities as defects can easily cause stress concentration, which deteriorates the mechanical properties of the WG/PU composites. The micro-structural analysis of WG/PU composites ensures the excellent emulsifying ability of the CTS, which is consistent with the results of mechanical performance testing.
3.5 IR spectra of WG/PU composites
The intensity of the NCO peak is a very important parameter, expressing the degree of curing of WG/PU composites. The strong absorption peak at 2270 cm−1 corresponds to the NCO group stretching in the WG/PU composites. As shown in Figure 6, whether modified or not, the NCO peak intensity of the WG/PU composites gradually decreases as the the curing time increases from 0.5 d to 28 d. This implies that NCO groups in the organic phase are gradually consumed by the diffused water from the inorganic phase during the curing process of the WG/PU composites. Moreover, by comparison, the NCO peak intensity of the modified WG/PU composites is weaker than that of the non-modified WG/PU composites under the same curing time. This difference indicates that the curing process of the WG/PU composites can be accelerated by the addition of CTS due to the intensity of the peak decreases progressively as the curing rate increases. In addition, in comparison with the non-modified WG/PU composites, the absorption peak of NCO groups exhibited by the modified WG/PU composites decreases after fully curing. This is due to the fact that the CTS enters the organic phase as the additional crosslinking points form chemical interactions with NCO groups in the organic phase.
The DRIFTS spectrum obtained for the modified WG/PU composites is compared with those for the non-modified WG/PU composites in Figure 7, which shows that the nearly identical spectrum curves of modified and non-modified WG/PU composites. This indicates that the addition of CTS has no obvious effect on the change of the chemical structure of the WG/PU composites. The absorption peaks at 3280-3360 cm-1 and 1680 cm-1 might contribute to amide N-H stretching peak and the urea carbonyl groups (C=O), which is the essential structures for polyurea from the reaction between NCO groups and water molecules. However, the characteristic peak of NCO at 2267 cm-1 is still observed, indicating that there might be a small amount of residual NCO in the WG/PU composites after fully curing.
3.6 Thermo gravimetric analysis of WG/PU composites
In order to fully evaluate the effect of CTS on thermal properties of WG/PU composites, we investigate the course of thermal decomposition of WG and WG/PU composites before and after modification (see Figure 8a). In the temperature range from 30 ºC to 250 ºC the free water and the crystallization water are lost from the WG, with maximu rate at 80 ºC and 132 ºC, resepcetively. Similarly, the DTG peaks of WG/PU composites within 50 ºC-250 ºC are due to elimination of water and a small amount of low molecular weight organics release. Between 50 ºC and 178 ºC, an obvious reduction in mass loss rate for modified WG/PU composites compares to non-modified WG/PU composites. It results from the fact the dispersion of WG particles in the PU matrix is higher and finer than in case of non-modified WG/PU compsites, therefore, the water from the WG can be more easily consumed. Moreover, a strong degradation peak is detected between 178 °C and 220 °C, which should be linked with the evaporation of the CTS present at the inorganic-organic phase interface of the modified WG/PU compsites. Besides, three other typical weight loss steps are observed: the first and second mass loss occurring at 330 °C and 412 °C can be attributed to the degradation of hard and soft segment, respectively21. The third weight loss in the range of 480 °C-550 °C might be due to the degradation of the rest of the organic phase22. In this step, the weight loss in the temperature range of 490 °C-506 °C seems to be ascribed to the decomposition of the CTS23. Moreover, the decomposition temperature is notably higher than the boiling point (195 °C) of the CTS becasue the CTS forms strong chemical bonds with the organic phase.
Figure 8b displays the DSC thermograms of the non-modified and modified WG/PU composites. The DSC thermograms shows that there is a complex structure in the WG/PU composites. It will not clearly discern the glass transition temperature (Tg) of the WG/PU composites. This phenomenon is attributed to that Tg is superimposed to thermal evaporation and degradation24. In the non-modified and modified WG/PU composites, the broad endothermic peak at 120 °C-210 °C is ascribed to the endothermic melting peak of hard segment (Tm, hs). The addition of CTS has a notable effect on the Tm, hs value of the WG/PU composites, which can be observed from the incremental change in Tm, hs values. It mainly attributed to the increasing of hard segments contents and crosslinking density by the CTS as the active hard segment corsslinker chemical links with the organic phase. A strong endothermic peak at 190 °C superimposes to the Tm, hs, and the result confirms that the evaporation of the CTS because this temperature is close to the boiling point of free CTS. It also indicates that partially CTS is not bonded to the dispersed or continuous phases in the WG/PU composite, which is in good agreement with the TGA results.
{kind=link}