Animals
Adult male (250–300 g) Wistar rats were used. Our experimental protocols were approved by the Division of Laboratory Animal Units, Cinvestav-IPN, and were in compliance with federal law and Consejo Nacional de Ciencia y Tecnología regulations.
Isolation of hearts
In preparation for heart extraction, each rat was anesthetized with 50 mg/kg sodium pentobarbital and given 500 U/kg heparin sodium solution (both administered by intraperitoneal injection). When the rat was completely unresponsive to stimulation, its heart was excised rapidly, arrested in modified Krebs-Henseleit buffer (containing, in mM: 117.8 NaCl, 1.2 NaH2PO4, 6.0 KCl, 24.3 NaHCO3, 1.2 MgSO4, 0.027 EDTA, 5.1 glucose and 1.6 CaCl2), gassed with 95% O2/5% CO2 at pH 7.4, and perfused in a Langendorff apparatus with an aortic cannula. Unless otherwise stated, all chemicals and materials were purchased from Sigma-Aldrich (St. Louis, MO).
Isolated hearts in the control group were perfused with Krebs-Henseleit buffer for 90 min. Those in the Dzx-treated (Tocris, Bristol, UK) group were perfused in Krebs-Henseleit buffer containing 100 mM Dzx for 90 min. Hearts in the NAC-Dzx group were first exposed to Krebs-Henseleit buffer with 4 mM N-acetyl cysteine (NAC), a reactive oxygen species (ROS) scavenger, for 15 min. Thereafter, the perfusion buffer was supplemented with 100 mM Dzx for 90 min. Hearts in the NAC group were first perfused with Krebs-Henseleit buffer for 90 min, to which NAC (4 mM) was added. For experiments in which the mKATP channel antagonist 5-hydroxydecanoate (5-HD) was used, 100 mM 5-HD was applied as described above for NAC.
Isolation of ventricular myocytes
Excised hearts were perfused for 5 min at 37 °C with Ca2+-free Tyrode’s solution containing (in mM): 136 NaCl, 5.4 KCl, 1 MgCl2, 10 HEPES, and 11 glucose. Hearts were recirculated for 60 min with Tyrode’s solution supplemented with 70-U/mL type II collagenase (Worthington, Lakewood, NJ) and 0.5-mg/100 mL type XIV protease. Ventricles were minced and shaken 2–3 times at 45 rpm for 7 minutes in the same solution. The dislodged cells were filtered through a 100-mm nylon cell strainer (BD Falcon) and centrifuged at 28 ×g for 2 min. The pellet was resuspended in Tyrode’s solution with 1% bovine serum albumin (BSA).
Cardiomyocyte treatments
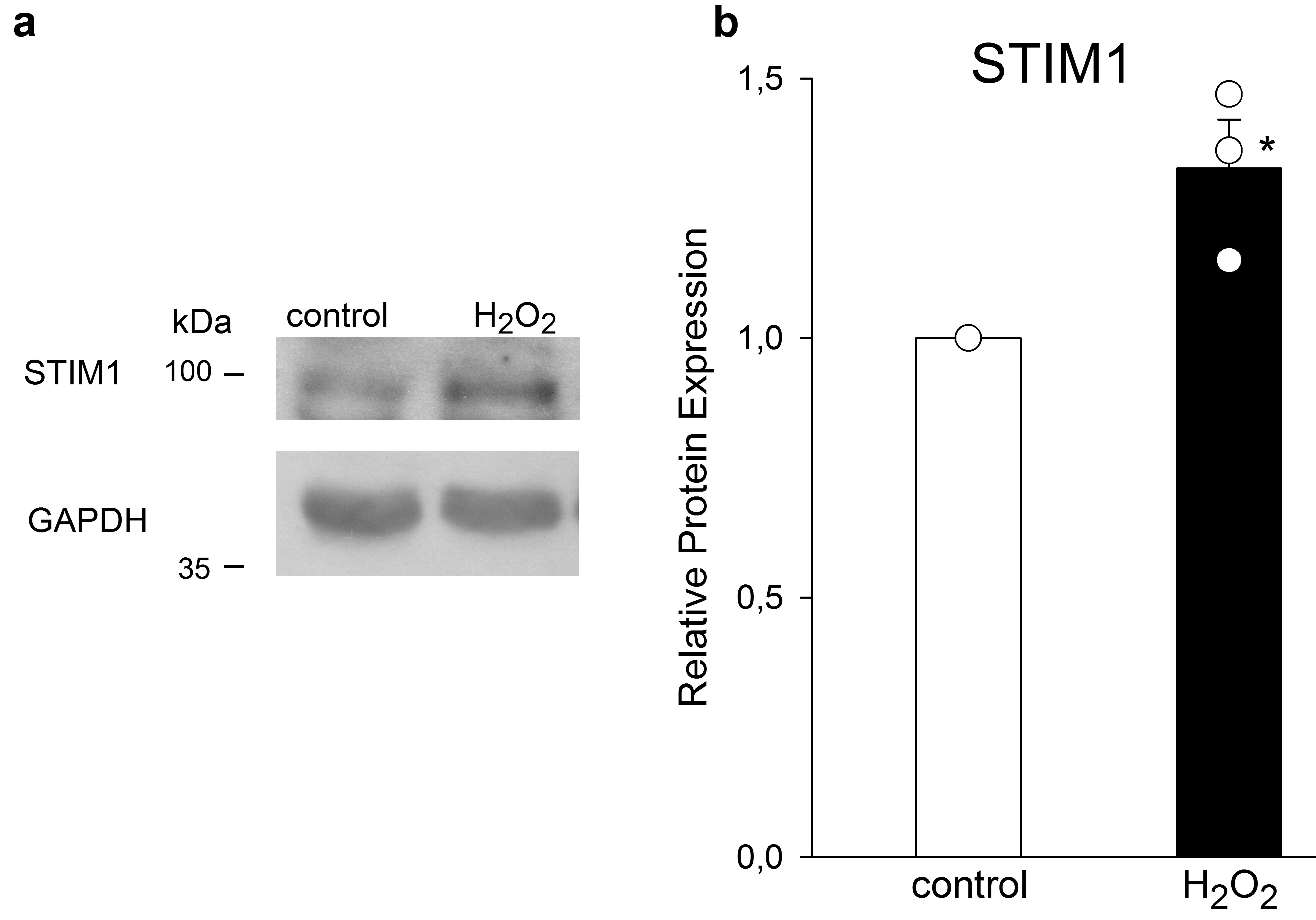
Resuspended pellets were maintained for 90 min in Tyrode solution plus 1% BSA in control experiments, or in an identical solution containing Dzx (100 mM), 5-HD (100 mM), or NAC (2 mM or 4 mM). To test the involvement of the MAPK pathway, we used 5 mM 1,4-diamino-2,3-dicyano-1,4-bis(methylthio) butadiene (UO126), a selective noncompetitive inhibitor of the MAPK kinases, MEK1 and MEK2. Cardiomyocytes were preincubated for 1 h in Tyrode solution containing UO126 and then Dzx was added to this solution and cardiomyocytes were incubated for additional 90 min. Cardiomyocytes were exposed to 10 mM cycloheximide (CHX), a selective inhibitor of protein synthesis, for 30 min and then incubated for 90 min in the same solution with 100 mM Dzx added. To test the involvement of ROS, we added 100 mM H2O2 to Tyrode solution for 10 min and then cardiomyocytes were incubated for additional 90 min in H2O2-free Tyrode solution.
All drugs were removed by washing three times with Tyrode’s solution containing BSA (1 mg/mL) and 1-mM CaCl2. Thereafter, cells were centrifuged at 28 ×g for 2 min, and total proteins were extracted for western blot analysis.
Membrane fractionation and western blotting
To obtain the membrane fraction, heart tissue was homogenized in ice-cold lysis buffer containing (in mM) 20 Tris (pH 7.4), 5.0 EDTA, 250 sucrose, 1.0 phenylmethanesulfonylfluoride, and 2.5% protease inhibitor mixture, as described elsewhere[19]. Tissue homogenates (20% w/v) were centrifuged at 1,000 ×g for 10 min to remove nuclei and debris, and the supernatant was ultracentrifuged at 110,000 ×g for 75 min at 4 °C to pellet the crude membrane fraction (sarcolemmal and microsomal subfractions). The resulting pellet was resuspended in solubilization buffer containing (in mM) 50 Tris (pH 7.4), 100 NaCl, 50 LiCl, 5 EDTA, 0.5% (v/v) Triton X-100, 0.5% (w/v) sodium deoxycholate, 0.05% (w/v) sodium dodecyl sulfate (SDS), and 0.02% (w/v) sodium azide. After incubation for 30 min on ice, the remaining insoluble material was collected by centrifugation (14,000 ×g, 10 min, 4 °C). Protein content of the supernatant (particulate membrane fraction) was measured with the Bradford method.
Dissociated myocytes used for western blotting were resuspended in lysis buffer containing 20 mM Tris (pH 7.5), 100 mM NaCl, 1% Triton X-100, and protease inhibitors. Lysis was achieved by vortexing every 10 min, for 60 min, at 4°C, followed by five cycles of sonication. Samples were centrifuged at 13,000 ×g for 10 min at 4°C and the soluble fraction was used for western blots. Protein content was measured with Bradford assays.
Whole-membrane fractions from ventricles or total fraction samples from isolated cardiomyocytes (50–60 mg) were subjected to 10% SDS-polyacrylamide gel electrophoresis (180 V, 120 min). The resultant protein bands were transferred onto nitrocellulose membranes, blocked with 4.5% nonfat dried milk in PBS, and probed with anti-STIM1 monoclonal antibody (1:1000; Abcam, Cambridge, UK), anti-Orai1 polyclonal antibody (1:3000; Abcam, Cambridge, UK), anti–phosphorylated-44/42 MAPK (pERK1/2) polyclonal antibody (1:1000; Cell Signaling Technology, Danvers, MA, USA) and anti–ERK1/2-44/42 MAPK (ERK1/2) monoclonal antibody (1:500; Santa Cruz Biotechnology Inc., Dallas, TX, USA). This antibody recognizes two bands of total ERK with molecular weights of 44 kDa (ERK1) and 42 kDa (ERK2), being the most abundant one the band of lower molecular weight. Finally, as loading controls we used anti-actin monoclonal antibody (1:2000; Sigma Aldrich, St. Louis, MO, USA) and anti-GAPDH monoclonal antibody (1:15,000; Sigma Aldrich, St. Louis, MO, USA) in PBS for 12–14 h at 4 °C. After incubation, membranes were rinsed three times with PBS-Tween20 (0.1%), incubated for 1 h with anti-rabbit (1:50,000) or anti-mouse (1:75,000) horseradish peroxidase-conjugated secondary antibody (Invitrogen, Carlsbad, CA, USA) in PBS and rinsed with PBS-Tween 20 (0.1%). Antibody labeling was detected with Immobilon western reagent (Millipore Co, Billerica, MA, USA) according to the manufacturer’s instructions.
Immunofluorescence
Freshly isolated adult rat cardiomyocytes were suspended in Tyrode’s solution containing 1 mM CaCl2 plus 5% fetal bovine serum (Thermofisher Scientific, Waltham, MA, USA), plated onto laminin-treated slides and allowed to settle for 2 h at 37 °C. Attached cardiomyocytes were then subjected to the aforementioned drug treatments. All drugs were removed by washing three times with Tyrode’s solution containing 1mM CaCl2. Immediately after the treatments, cells were fixed with 4% paraformaldehyde in PBS for 15 min at 4 °C. The fixed cells were washed three times with PBS, and then permeabilized and blocked in PBS containing 0.3 % Triton X-100 and 5% donkey serum for 1 h at room temperature. Cells were then incubated overnight at 4 °C with primary antibodies: monoclonal anti-STIM1 (1:50; Abcam, Cambridge, UK), polyclonal anti-Orai1 (1:50; Thermofisher Scientific, Waltham, MA, USA), monoclonal anti c-Fos (1:50, Santa Cruz), monoclonal anti NFkBp65 (1:50; Santa Cruz, CA, USA) in PBS containing 0.3% Triton X-100 and 0.5% BSA. After washing three times in PBS, cells were incubated for 1 h at room temperature with secondary antibodies: Alexa Fluor 555-conjugated donkey anti-rabbit (1:200; Thermofisher Scientific, Waltham, MA, USA) for Stim1 and Alexa Fluor 488-conjugated donkey anti-mouse (1:200; Thermofisher Scientific, Waltham, MA, USA) for Orai1 and then washed three times in PBS. Thereafter, cells were incubated for 10 min at room temperature with Hoechst nuclear stain (1:1000; Invitrogen, Carlsbad, CA, USA). Negative controls were processed without primary antibodies.
Labeling was visualized under a laser scanning confocal microscope (Leica, Wetzlar, Germany, model TCS-SP8) with argon (488 nm) and helium/neon (543 nm) lasers used with an optimized pinhole diameter. Confocal images were obtained as z-stacks of single optical sections, which were superimposed as a single image in Leica LAS AF 2.6.0 build 7268 software. Immunofluorescence was quantified in ImageJ 1.44 p (NIH, Bethesda, MD, USA) after the images were threshold adjusted (pixel value used to find edges of immunolabeling closed regions) at an intensity of twice the mean intensity and three (for STIM1) or five (for Orai1) times the standard deviation. The threshold area was outlined under particle analysis (size 0-infinity; circularity 0-1).
qRT-PCR
RNA was isolated from cardiomyocytes with RLT buffer and a RNeasy Mini Kit (Qiagen, Hilden, Germany), and cDNAs were synthesized with a Taqman reverse transcription kit (Thermofischer, Waltham, MA, USA). Transcript levels were determined in TaqMan assays (Rn02397170_m1, Rn01506496_m1; Applied Biosystems, Foster City, CA, USA) and an iCycler iQ machine (Bio-Rad, Hercules, CA) with TaqMan Gene Expression Master Mix (Thermofischer, Waltham, MA, USA). mRNA expression was assessed relative to the ribosomal RNA 18S (Hs999999_s1, Applied Biosystems, Foster City, CA, USA), as recommended by the manufacturer. Relative changes in expression were calculated by the 2-DDCT method[20].
Data analysis
The data, which are expressed as means ± standard errors of the mean (SEMs), were tested for normal distribution, analyzed with independent t-tests (when two groups were compared) or with analyses of variance (ANOVAs) followed by multiple comparison Dunnett’s tests (each treatment group vs. control group). A significance criterion of p < 0.05 was used.
{kind=link}