Chemistry. All solvents were dried and purified prior to use according to standard procedures. When required, reactions were performed under inert atmosphere (Ar) in preflamed glassware. Anhydrous Na2SO4 was used for drying solutions, and the solvents were then routinely removed at ca. 40 oC under reduced pressure using a rotary vacuum evaporator. All reagents employed in the present work were commercially available and used without further purification. Among all the amines employed in the synthesis of the betulin carbamates, 6a-m and tert-butyl-4-(2-aminoethyl) piperazine-1-carboxylate, used for the synthesis of 6j, were prepared according to a previously published protocol 43. 2-(3,4-dimethoxyphenyl)ethan-1-amine and 2-(3,4,5-trimethoxyphenyl)ethan-1-amine employed in the synthesis of 6f and 6g, respectively, were synthesized and characterized according to the procedures described in Supplementary Material. Flash column chromatography (FCC) was performed on silica gel (70–230 and 230–400 mesh, Merck, Germany) and analytical thin layer chromatography (TLC) on silica 60gel-F254 precoated aluminum foils (0.2 mm film, Merck, Germany). Spots on the TLC plates were visualized with UV light at 254 nm and using anisaldehyde solution. 1H NMR spectra were recorded in CDCl3 at 600.13 MHz and 13C spectra at 150.9 MHz on a Bruker AVANCEIII HD spectrometer. Chemical shifts (δ) are indicated in parts per million downfield from TMS. Coupling constants (J) are reported in Hertz. HR mass spectra were performed using an ESI-LTQ-ORBITRAP XL unit (Thermo Scientific, Bremen, Germany). The Orbitrap Unit was operated in positive mode, with a spray voltage of 3.2 kV, while the sheath gas flow rate and auxiliary gas flow rate were adjusted to 12 and 2 arbitrary units, respectively. The capillary voltage and the tube lens voltage were set to 10 and 110 V, respectively. The scan ranged from m/z 150 up to 2000. The purity of the tested compounds was determined by reversed phase high performance liquid chromatography (HPLC) (Jasco LC-NetII/ADC series) equipped with a Phenomenex Gemini-NX RP-C18 150 x 4.6 mm, 5 µm particle size column and a MD-4010 PDA Detector and monitored at 254 and 280 nm. Linear gradients of A and B were used where A = 0.1% TFA in H2O and B = 0.1% TFA in CH3CN, at a flow rate of 0.8 mL/min. The purity of the tested compounds, which corresponded to > 95% (see the Supplementary Material), was evaluated as a percentage ratio between the areas of the main peak and of possible impurities.
General procedure for the synthesis
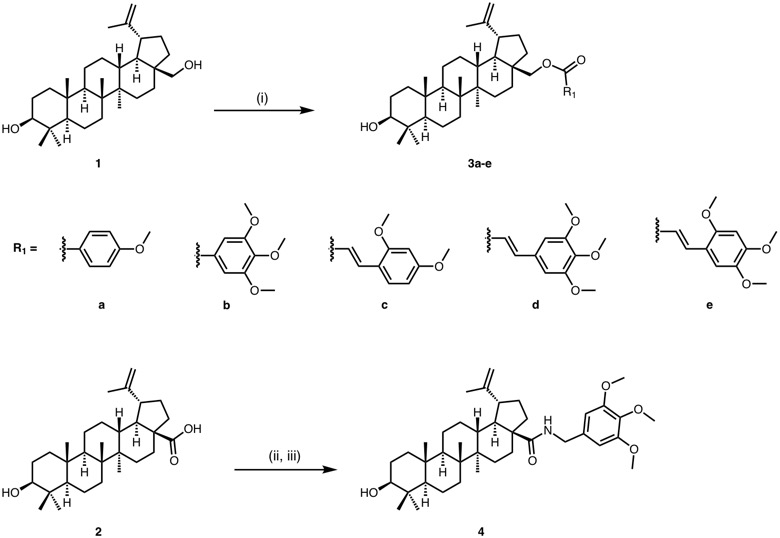
General procedure for the synthesis of betulin esters 3a-e.
To a stirring solution of compound 1 in dry DCM/THF (3:2 v/v, 0.05 M), under Ar atmosphere, the aromatic acid (1.5 eq) and 4-dimethylaminopyridine (1.75 eq) were added, and the suspension was cooled to 0°C. Subsequently DCC (1.75 eq) was added, and the reaction mixture was stirred for 5 min at 0°C and then at ambient temperature overnight. Upon consumption of the starting material, dicyclohexylurea was filtered under vacuum and washed in the filtration funnel with ethyl acetate (AcOEt). After evaporation of the solvent to dryness under reduced pressure, the residue was subjected to FCC, affording the pure betulin esters 3a-e as colorless oils.
3a: compound 1 was reacted with 4-methoxybenzoic acid according to the general procedure. Yield = 98%; Colorless oil; Rf (PhMe/EtOAc 95:5) = 0.27; 1H NMR (600 MHz, CDCl3) δ 8.01 (br s, 1 H), 7.99 (br s, 1 H), 6.93 (br s, 1 H), 6.91 (br s, 1 H), 4.72 (d, J = 2.5 Hz, 1 H), 4.60 (t, J = 1.4 Hz, 1 H), 4.48 (dd, J = 11.1, 2.1 Hz, 1 H), 4.06 (dd, J = 11.0, 1.4 Hz, 1 H), 3.86 (s, 3 H), 3.18 (dd, J = 11.5, 4.8 Hz, 1 H), 2.52 (td, J = 11.3, 5.9 Hz, 1 H), 2.06–1.95 (m, 2 H), 1.91 (ddd, J = 12.4, 8.4, 1.2 Hz, 1 H), 1.80–1.73 (m, 2 H), 1.70 (s, 3 H), 1.68–1.21 (m, 16 H), 1.18–1.08 (m, 2 H), 1.06 (s, 3 H), 1.0 (s, 3 H), 0.97 (s, 3 H), 0.91 (td, J = 14.1, 4.4 Hz, 1 H), 0.83 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.5 Hz,1 H); 13C NMR (151 MHz, CDCl3) δ 166.7, 163.3, 150.2, 131.5, 122.9, 113.6, 109.8, 79.0, 62.9, 55.4, 55.3, 50.4, 48.9, 47.8, 46.7, 42.8, 40.9, 38.9, 38.7, 37.6, 37.2, 34.7, 34.2, 30.0, 29.7, 28.0, 27.4, 27.2, 25.2, 20.8, 19.2, 18.3, 16.1, 16.1, 15.3, 14.8; HR-ESI (m/z) [2M + H+] calculated for C38H57O4 1153.8438. Found: 1153.8428; HPLC analysis: peak area 99.5%, tR=14.30 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
3b: compound 1 was reacted with 3,4,5-trimethoxybenzoic acid according to the general procedure. Yield = 71%; Colorless oil; Rf (PhMe/EtOAc 9:1) = 0.24; 1H NMR (600 MHz, CDCl3) δ 7.31 (s, 2 H), 4.72 (d, J = 2.6 Hz, 1 H), 4.61 (t, J = 1.5 Hz, 1 H), 4.53 (dd, J = 11.1, 1.9 Hz, 1 H), 4.08 (d, J = 11.1 Hz, 1 H), 3.91 (s, 9 H), 3.19 (dd, J = 11.6, 4.8 Hz, 1 H), 2.54 (td, J = 11.1, 5.8 Hz, 1 H), 2.08-2.00 (m, 1 H), 1.95–1.86 (m, 2 H), 1.81–1.72 (m, 2 H), 1.71 (s, 3 H), 1.69–1.65 (m, 2 H), 1.64–1.14 (m, 16 H), 1.07 (s, 3 H), 1.00 (s, 3 H), 0.97 (s, 3 H), 0.91 (td, J = 13.4, 4.4 Hz, 1 H), 0.83 (s, 3 H), 0.76 (s, 3 H), 0.69 (d, J = 9.4 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 166.5, 152.9, 150.1, 125.5, 110.0, 106.8, 79.0, 63.5, 60.9, 56.3, 55.3, 50.4, 48.8, 47.8, 46.8, 42.8, 40.9, 38.9, 38.7, 37.7, 37.2, 34.8, 34.2, 30.1, 29.7, 28.0, 27.4, 27.2, 25.2, 20.8, 19.1, 18.3, 16.1, 16.0, 15.3, 14.8; HR-ESI (m/z) [2M + H+] calculated for C40H61O6 1273.8860. Found: 1273.8845; HPLC analysis: peak area 99.3%, tR=12.90 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
3c: compound 1 was reacted with trans-2,4-dimethoxycinnamic acid according to the general procedure. Yield = 95%; Colorless oil; Rf (PhMe/EtOAc 9:1) = 0.4; 1H NMR (600 MHz, CDCl3) δ 7.92 (d, J = 16.2 Hz, 1 H), 7.45 (d, J = 8.6 Hz, 1 H), 6.50 (dd, J = 8.5, 2.4 Hz, 1 H), 6.45–6.42 (m, 2 H), 4.71 (d, J = 2.5 Hz, 1 H), 4.60 (t, J = 1.5 Hz, 1 H), 4.39 (dd, J = 11.1, 2.0 Hz, 1 H), 3.97 (d, J = 11.1 Hz, 1 H), 3.87 (s, 3 H), 3.84 (s, 3 H), 3.18 (dd, J = 11.4, 4.8 Hz, 1 H), 2.50 (td, J = 11.2, 5.9 Hz, 1 H), 2.05–1.98 (m, 1 H), 1.93 (m, 1 H), 1.89–1.84 (m, 1 H), 1.79–1.71 (m, 2 H), 1.69 (s, 3 H), 1.68–1.39 (m, 13 H), 1.30–1.23 (m, 3 H), 1.22–1.08 (m, 2 H), 1.06 (s, 3 H), 0.99 (s, 3 H), 0.97 (s, 3 H), 0.90 (td, J = 12.6, 4.0 Hz, 1 H), 0.83 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.6 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 168.4, 162.7, 159.8, 150.3, 140.0, 130.4, 116.7, 116.1, 109.8, 105.2, 98.4, 79.0, 62.5, 55.4, 55.3, 50.4, 48.9, 47.7, 46.6, 42.7, 40.9, 38.9, 38.7, 37.6, 37.2, 34.7, 34.2, 29.9, 29.7, 28.0, 27.4, 27.2, 25.2, 20.8, 19.2, 18.3, 16.1, 16.0, 15.3, 14.8; HR-ESI (m/z) [M + H+] calculated for C41H61O5 633.4519. Found: 633.4516; HPLC analysis: peak area 94.8%, tR=13.20 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
3d: compound 1 was reacted with 3,4,5-trimethoxycinnamic acid according to the general procedure. Yield = 64%; Colorless oil; Rf (PhMe/EtOAc 9:1) = 0.29; 1H NMR (600 MHz, CDCl3) δ 7.59 (d, J = 15.6 Hz, 1 H), 6.75 (s, 2 H), 6.36 (d, J = 15.6 Hz, 1 H), 4.71 (d, J = 2.5 Hz, 1 H), 4.60 (t, J = 1.5 Hz, 1 H), 4.40 (dd, J = 11.4, 2.0 Hz, 1 H), 4.00 (d, J = 11.4 Hz, 1 H), 3.89 (s, 6 H), 3.88 (s, 3 H), 3.18 (dd, J = 11.4, 4.8 Hz, 1 H), 2.50 (td, J = 11.1, 5.6 Hz, 1 H), 2.05–1.97 (m, 1 H), 1.96–1.91 (m, 1 H), 1.89–1.83 (m, 1 H), 1.78–1.71 (m, 2 H), 1.70 (s, 3 H), 1.65–1.24 (m, 18 H), 1.06 (s, 3 H), 1.00 (s, 3 H), 0.97 (s, 3 H), 0.91 (td, J = 13.1, 4.2 Hz, 1 H), 0.83 (s, 3 H), 0.76 (s, 3 H), 0.69 (d, J = 11.2 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 167.4, 153.4, 150.1, 144.6, 140.1, 129.9, 129.0, 128.2, 117.5, 109.9, 105.2, 79.0, 62.9, 61.0, 56.2, 55.3, 50.4, 48.9, 47.7, 46.6, 42.7, 40.9, 38.9, 38.7, 37.6, 37.2, 34.7, 34.2, 29.9, 29.6, 28.0, 27.4, 27.1, 25.2, 20.8, 19.1, 18.3, 16.1, 16.0, 15.3, 15.2, 14.8; HR-ESI (m/z) [M + H+] calculated for C42H63O6 663.4624. Found: 633.4570; HPLC analysis: peak area 99.1%, tR=12.60 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
3e: compound 1 was reacted with 2,4,5-trimethoxycinnamic acid according to the general procedure. Yield = 47%; Colorless oil; Rf (PhMe/EtOAc 9:1) = 0.16; 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 16.2 Hz, 1 H), 7.02 (s, 1 H), 6.50 (s, 1 H), 6.38 (d, J = 16.2 Hz, 1 H), 4.71–4.70 (m, 1 H), 4.60 (t, J = 1.6 Hz, 1 H), 4.40 (dd, J = 11.2, 1.9 Hz, 1 H), 3.98 (d, J = 11.4 Hz, 1 H), 3.93 (s, 3 H), 3.87 (s, 3 H), 3.86 (s, 3 H), 3.19 (dd, J = 11.4, 4.8 Hz, 1 H), 2.51 (td, J = 11.1, 5.7 Hz, 1 H), 2.07–1.98 (m, 1 H), 1.96–1.91 (m, 1 H), 1.89–1.85 (m, 1 H), 1.80–1.71 (m, 2 H), 1.70 (s, 3 H), 1.66–1.11 (m, 18 H), 1.06 (s, 3 H), 0.99 (s, 3 H), 0.97 (s, 3 H), 0.91 − 0.86 (m, 1 H), 0.83 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.5 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 168.2, 153.9, 152.1, 150.3, 143.3, 139.5, 129.0, 125.3, 115.8, 115.1, 110.9, 109.8, 96.9, 79.0, 62.5, 56.5, 56.0, 55.3, 50.4, 48.9, 47.7, 46.6, 42.7, 40.9, 38.9, 38.7, 37.6, 37.2, 34.7, 34.2, 29.9, 29.7, 28.0, 27.4, 27.2, 25.2, 20.8, 19.1, 18.3, 16.1, 16.0, 15.3, 14.8; HR-ESI (m/z) [M + H+] calculated for C42H63O6 663.4624. Found: 633.4619; HPLC analysis: peak area 98.1%, tR=11.33 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
General procedure for the synthesis of amide 4.
To an ice-cold stirring solution of compound 2 (20 mg, 0.044 mmol) in dry THF, HOBt (8 mg, 0.06 mmol) and DCC (10 mg, 0.05 mmol) were added, and the solution was stirred at room temperature for 15 h. After completion of the reaction, the mixture was diluted in 1,2-dichloroethane (DCE) and washed sequentially with 5% aqueous ice-cold NaHCO3, water and brine. Then it was dried over anhydrous Na2SO4, filtered and concentrated to dryness, to provide the corresponding active ester as a white solid (22mg, 88% yield); 1H NMR (600 MHz, CDCl3) δ 8.08 (d, J = 8.4 Hz, 1 H), 7.56 (ddd, J = 7.2, 1.2, 0.6 Hz, 1 H), 7.43 (ddd, J = 7.2, 1.2, 0.6 Hz, 1 H), 7.36 (d, J = 8.2 Hz, 1 H), 4.73 (d, J = 2.3 Hz, 1 H), 4.63 (t, J = 1.7 Hz, 1 H), 4.17 (d, J = 8.4 Hz, 1 H), 3.51–3.43 (m, 1 H), 3.21–3.15 (m, 2 H), 2.95 (td, J = 11.2, 5.0 Hz, 1 H), 2.65–2.61 (m, 1 H), 2.41 (dd, J = 13.0, 8.1 Hz, 1 H), 2.21 (td, J = 12.6, 3.7 Hz, 1 H), 2.12–2.04 (m, 1 H), 1.71 (s, 3 H), 1.61–1.51 (m, 7 H), 1.47–1.40 (m, 4 H), 1.35–1.25 (m, 6 H), 1.18–1.24 (m, 2 H), 1.08–1.10 (m, 1 H), 1.04 (s, 3 H), 0.98 (s, 3 H), 0.97 (s, 3 H), 0.90 (td, J = 13.6, 4.1 Hz, 1 H), 0.81 (s, 3 H), 0.76 (s, 3 H), 0.69–0.72 (m, 1 H); 13C NMR (151 MHz, CDCl3) δ 171.8, 157.1, 149.2, 143.6, 128.9, 128.7, 124.7, 120.6, 110.4, 107.9, 78.9, 57.0, 55.8, 55.4, 50.5, 49.9, 49.1, 46.6, 42.5, 40.8, 38.9, 38.7, 38.45, 37.2, 36.8, 34.9, 34.4, 33.9, 31.4, 30.3, 30.1, 28.0, 27.4, 25.6, 25.4, 24.9, 20.8, 19.4, 18.3, 16.1, 15.4, 14.8.
To a solution of the above active ester (22 mg, 0.04 mmol) in dry THF (50 µL) under Ar atmosphere and stirring at 0°C, 3,4,5-trimethoxybenzylamine (12 µL, 0.067 mmol) was added and the reaction mixture was stirred for another 16 h at ambient temperature. After completion of the reaction, the mixture was diluted in ethyl acetate and washed sequentially with 5% aqueous ice-cold citric acid, water, and brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The residue obtained was subjected to FCC using PhMe/EtOAc 9:1 as eluent, affording the corresponding amide 4 in 50% yield; White solid; m.p.: 99.7-100.5°C; Rf (PhMe/EtOAc 95:5) = 0.08; 1H NMR (600 MHz, CDCl3) δ 6.49 (s, 2 H), 5.94–5.89 (m, 1 H), 4.74 (d, J = 2.9 Hz, 1 H), 4.60 (t, J = 2.2 Hz, 1 H), 4.40 (dd, J = 14.4, 5.8 Hz, 1 H), 4.34 (dd, J = 14.8 Hz, 5.8 Hz, 1 H), 3.84 (d, J = 1.2 Hz, 6 H), 3.83 (d, J = 0.6 Hz, 3 H), 3.19–3.12 (m, 2 H), 2.52 (td, J = 13.0, 3.7 Hz, 1 H), 1.97–1.90 (m, 2 H), 1.78 (dd, J = 12.3, 7.6 Hz, 1 H), 1.73–1.71 (m, 1 H), 1.69 (s, 3 H), 1.66–1.64 (m, 1H), 1.62–1.52 (m, 5 H), 1.58–1.33 (m, 9 H), 1.30–1.23 (m, 3 H), 1.14 (dt, J = 13.3, 3.0 Hz, 1 H), 0.97 (s, 3 H), 0.96 (s, 3 H), 0.95 − 0.92 (m, 1 H), 0.90 (s, 3 H), 0.81 (s, 3 H), 0.75 (s, 3 H), 0.68 − 0.65 (m, 1 H); 13C NMR (151 MHz, CDCl3) δ 175.9, 153.4, 150.8, 137.1, 135.0, 129.0, 128.2, 109.5, 104.6, 79.0, 60.8, 56.0, 55.7, 55.4, 50.6, 50.1, 46.7, 43.3, 42.5, 40.7, 38.8, 38.7, 38.4, 37.6, 37.2, 34.4, 33.7, 30.9, 29.4, 28.0, 27.4, 25.6, 24.9, 20.9, 19.5, 18.3, 16.2, 16.1, 15.3, 14.6; HR-ESI (m/z) [M + H+] calculated for C40H61NO5 635.4550. Found: 635.4573; HPLC analysis: analysis: peak area 98.9%, tR=6.02 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
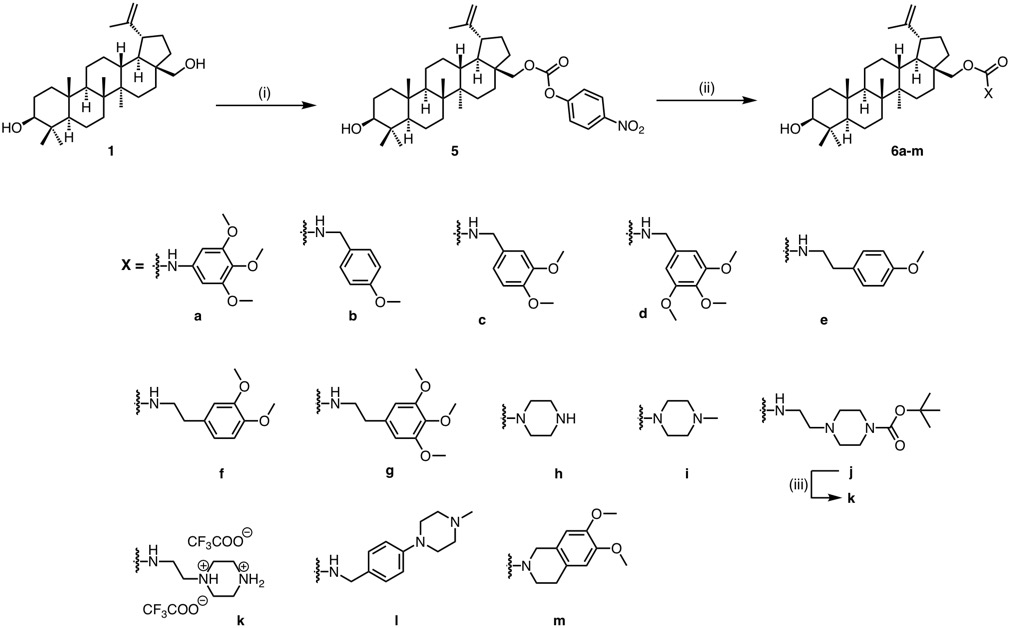
General procedure for the synthesis of betulin p -nitrophenylcarbonate ester 5
To a stirring solution of compound 1 (200 mg, 0.45 mmol) in dry THF (10 mL), under Ar atmosphere, pyridine (36 µL, 0.45 mmol) was added, and the mixture was cooled to 0 oC. p-nitrophenylchloroformate (94.3 mg, 0.47 mmol) was added in 3 portions and the reaction mixture was stirred for 24 h at ambient temperature. Consequently, the mixture was concentrated under reduced pressure to dryness. The solid residue obtained was diluted in DCM and washed sequentially with 5% aqueous ice-cold citric acid, water, and brine, dried over anhydrous Na2SO4, filtered and concentrated to dryness. The desired product was afforded as a white foam (227 mg) after FCC purification using PhMe/EtOAc 95:5 as eluent. Yield = 83%; White foam; Rf (PhMe/EtOAc 95:5) = 0.14; 1H NMR (600 MHz, CDCl3) δ 8.29 (s, 1 H), 8.28 (s, 1 H), 7.41 (s, 1 H), 7.39 (s, 1 H), 4.71 (br s, 1 H), 4.61 (t, J = 1.7 Hz, 1 H), 4.51 (dd, J = 10.8 Hz, 2.0 Hz, 1 H), 4.08 (d, J = 10.7 Hz, 1 H), 3.19 (dd, J = 11.4, 4.8 Hz, 1 H), 2.45 (td, J = 10.8, 5.4 Hz, 1 H), 2.03 (dtd, J = 14.2, 10.5, 4.9 Hz, 1 H), 1.95–1.84 (m, 2 H), 1.75 (td, J = 13.9, 4.7 Hz, 1 H), 1.70 (s, 3 H), 1.68–1.08 (m, 19 H), 1.05 (s, 3 H), 1.00 (s, 3 H), 0.97 (s, 3 H), 0.91 (td, J = 12.5, 3.7 Hz, 1 H), 0.83 (s, 3 H), 0.76 (s, 3 H), 0.69 (d, J = 11.2 Hz, 1 H);. 13C NMR (151 MHz, CDCl3) δ 155.6, 153.0, 149.7, 145.3, 125.3, 121.8, 78.9, 68.3, 55.3, 50.3, 48.8, 47.7, 46.7, 42.7, 40.9, 38.9, 38.7, 37.7, 37.1, 34.4, 34.2, 29.6, 29.5, 28.0, 27.4, 27.0, 25.2, 20.7, 18.3, 16.1, 16.0, 15.3, 14.8.
General procedure for the synthesis of betulin carbamates 6a-m.
To a stirring solution of carbonate ester 5 (0.16 mmol) in dry THF (0.2 M), under Ar atmosphere, the corresponding amine (appropriate eq for each case, see below) and DIPEA (1 eq) were added, and the reaction mixture was stirred at ambient temperature or heated at 50–60°C. After completion of the reaction the mixture was diluted in EtOAc and washed sequentially with 5% aqueous ice-cold citric acid, water, and brine, dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The residue thus obtained was purified by FCC to provide, upon solvent removal, the corresponding carbamates 6a-m.
6a: compound 6a was prepared using 3 equivalents of 3,4,5-trimethoxyaniline and overnight heating. Yield = 45%; Colorless oil; Rf (PhMe/EtOAc 9:1) = 0.11; 1H NMR (600 MHz, CDCl3) δ 6.69 (s, 1 H), 6.58 (s, 1 H), 4.70 (d, J = 2.5 Hz, 1 H), 4.60 (t, J = 1.7 Hz, 1 H), 4.34 (d, J = 10.7 Hz, 1 H), 4.07 (t, J = 6.8 Hz, 1 H), 3.98 (d, J = 10.1 Hz, 1 H), 3.85 (s, 6 H), 3.81 (s, 3 H), 3.19 (dd, J = 11.5, 4.7 Hz, 1 H), 2.47 (td, J = 11.1, 5.7 Hz, 1 H), 2.02–1.98 (m, 1 H), 1.91–1.71 (m, 4 H), 1.70 (s, 3 H), 1.66–1.51 (m, 5 H), 1.45–1.37 (m, 6 H), 1.30–1.25 (m, 2 H), 1.22 (dd, J = 12.8, 4.7 Hz, 1 H), 1.12–1.07 (m, 2 H), 1.06 (s, 3 H), 1.04-1.00 (m, 1 H), 0.99 (s, 3 H), 0.98 (s, 3 H), 0.95 − 0.92 (m, 1 H), 0.91 − 0.88 (m, 1 H), 0.83 (s, 3 H), 0.77 (s, 3 H), 0.69 (d, J = 9.5 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 153.4, 150.1, 134.0, 125.3, 121.8, 109.9, 79.0, 64.4, 61.0, 56.1, 55.3, 50.4, 48.8, 47.7, 46.5, 42.7, 40.9, 38.9, 38.7, 37.6, 37.1, 34.5, 34.2, 29.8, 29.6, 28.0, 27.4, 27.1, 25.2, 20.8, 18.3, 16.1, 15.3, 14.8, 13.7; HR-ESI (m/z) [M + H+] calculated for C40H62NO6 652.4509. Found: 652.4557; HPLC analysis: peak area 96.2%, tR=9.59 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
6b: compound 6b was prepared using 1.1 equivalents of 4-methoxybenzylamine at room temperature for 24 h. Yield = 60%; Colorless oil; Rf (PhMe/EtOAc 9:1) = 0.15; 1H NMR (600 MHz, CDCl3) δ 7.24–7.20 (m, 2 H), 6.88–6.84 (m, 2 H), 4.89 (br s, 1 H), 4.68 (d, J = 2.5 Hz, 1 H), 4.58 (t, J = 1.5 Hz, 1 H), 4.34–4.25 (m, 3 H), 3.88 (d, J = 10.6 Hz, 1 H), 3.80 (s, 3 H), 3.18 (dd, J = 11.5, 4.7 Hz, 1 H), 2.45 (tt, J = 10.8, 5 Hz, 1 H), 1.99 (h, J = 9.9 Hz, 2 H), 1.81 (d, J = 13.0 Hz, 1 H), 1.79–1.72 (m, 2 H), 1.71–1.69 (m, 1 H), 1.68 (s, 3 H), 1.62–1.50 (m, 9 H), 1.43–1.35 (m, 5 H), 1.26 (s, 3 H), 1.21 (dd, J = 13.2, 4.9 Hz, 2 H), 1.11 (s, 1 H), 1.04 (s, 3 H), 0.97 (s, 3 H), 0.93 − 0.86 (m, 1 H), 0.83 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.4 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 129.0, 128.2, 114.1, 109.8, 79.0, 55.3, 50.4, 48.8, 47.7, 46.6, 42.7, 40.9, 38.9, 38.7, 37.5, 37.1, 34.2, 29.7, 28.0, 27.4, 27.1, 25.2, 20.8, 18.3, 16.1, 16.1, 15.3, 14.7; HR-ESI (m/z) [M + H+] calculated for C39H60NO4 606.4522. Found: 606.4520; HPLC analysis: peak area 100%, tR=8.72 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
6c: compound 6c was prepared using 1.1 equivalents of 3,4-dimethoxybenzylamine at room temperature for 1 h. Yield = 66%; Colorless oil; Rf (PhMe/EtOAc 9:1) = 0.09; 1H NMR (600 MHz, CDCl3) δ 6.84 (s, 1 H), 6.82 (s, 2 H), 4.92 (s, 1 H), 4.69 (d, J = 2.5 Hz, 1 H), 4.58 (t, J = 1.5 Hz, 1 H), 4.31 (s, 2 H), 4.29 (s, 1 H), 3.87 (s, 3 H), 3.87 (s, 3 H), 3.18 (dd, J = 11.4, 4.8 Hz, 1 H), 2.45 (td, J = 11.1, 5.7 Hz, 1 H), 1.99 (qui, J = 9.9 Hz, 1 H), 1.82 (d, J = 13.7 Hz, 1 H), 1.79–1.72 (m, 3 H), 1.68 (s, 3 H), 1.66–1.11 (m, 19 H), 1.04 (s, 3 H), 0.97 (s, 6 H), 0.92 − 0.86 (m, 1 H), 0.82 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 8.4 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 149.2, 137.9, 129.0, 128.2, 125.3, 111.2, 111.0, 109.8, 79.0, 56.0, 55.9, 55.3, 50.4, 48.8, 47.7, 46.6, 42.7, 40.9, 38.8, 38.7, 37.5, 34.5, 34.2, 29.8, 29.6, 28.0, 27.4, 27.1, 25.2, 21.4, 20.8, 19.1, 18.3, 16.1, 16.0, 15.3, 14.8; HR-ESI (m/z) [M + H+] calculated for C40H62NO5 636.4628. Found: 636.4631; HPLC analysis: peak area 95.1%, tR=8.56 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
6d: compound 6d was prepared using 1.1 equivalents of 3,4,5-trimethoxybenzylamine at room temperature for 1.5 h. Yield = 62%; White solid; m.p.: 153.154.5 oC; Rf (PhMe/EtOAc 9:1) = 0.14; 1H NMR (600 MHz, CDCl3) δ 6.52 (s, 2 H), 4.96 (s, 1 H), 4.69 (d, J = 2.3 Hz, 1 H), 4.59 (t, J = 1.5 Hz, 1 H), 4.33 (s, 1 H), 4.31 (s, 2 H), 3.90 (d, J = 11.4 Hz, 1 H), 3.85 (s, 6 H), 3.83 (s, 3 H), 3.18 (dd, J = 11.4, 4.7 Hz, 1 H), 2.45 (td, J = 11.1, 5.8 Hz, 1 H), 2.04–1.94 (m, 1 H), 1.82 (d, J = 14.0 Hz, 1 H), 1.79–1.72 (m, 3 H), 1.68 (s, 3 H), 1.61–1.47 (m, 9 H), 1.44–1.34 (m, 5 H), 1.29–1.18 (m, 4 H), 1.05 (s, 3 H), 0.97 (s, 6 H), 0.90 (dd, J = 12.7, 3.7 Hz, 1 H), 0.82 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.2 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 153.4, 134.3, 109.8, 104.6, 79.0, 60.8, 56.1, 55.3, 50.4, 48.8, 47.7, 46.6, 42.7, 40.9, 38.9, 38.7, 37.5, 37.1, 34.5, 34.2, 29.8, 29.6, 28.0, 27.4, 27.1, 25.2, 20.8, 19.1, 18.3, 16.1, 16.0, 15.3, 14.8; HR-ESI (m/z) [M + H+] calculated for C41H64NO6 666.4733. Found: 666.4730; HPLC analysis: peak area 95.0%, tR=8.49 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
6e: compound 6e was prepared using 1.1 equivalents of 4-methoxyphenylethylamine at room temperature for 24 h. Yield = 55%; Colorless oil; Rf (PhMe/EtOAc 95:5) = 0.1; 1H NMR (600 MHz, CDCl3) δ 7.10 (d, J = 8.5 Hz, 2 H), 6.84 (d, J = 8.5 Hz, 2 H), 4.69 (d, J = 2.3 Hz, 1 H), 4.65 (br s, 1 H), 4.58 (t, J = 1.7 Hz, 1 H), 4.25 (dd, J = 10.9, 1.9 Hz, 1 H), 3.83 (d, J = 11.6 Hz, 1 H), 3.79 (s, 3 H), 3.45–3.37 (m, 2 H), 3.18 (dd, J = 11.5, 4.8 Hz, 1 H), 2.76 (t, J = 6.8 Hz, 2 H), 2.54–2.37 (m, 1 H), 1.97 (m, 1 H), 1.88–1.70 (m, 3 H), 1.68 (s, 3 H), 1.66–1.48 (m, 7 H), 1.44–1.35 (m, 5 H), 1.29–1.77 (m, 5 H), 1.11 (s, 1 H), 1.04 (s, 3 H), 1.02 (s, 1 H), 0.97 (m, 6 H), 0.91 (dd, J = 12.9, 3.7 Hz, 1 H), 0.82 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.6 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 158.3, 157.0, 150.5, 150.3, 129.7, 129.0, 128.2, 125.3, 114.0, 109.7, 79.0, 63.0, 60.5, 55.3, 50.4, 48.8, 47.8, 47.7, 46.6, 42.7, 40.9, 38.9, 38.7, 37.5, 37.1, 34.2, 29.7, 29.2, 28.0, 27.4, 27.1, 25.2, 21.4, 20.8, 19.1, 18.3, 16.1, 16.0, 15.3, 14.7; HR-ESI (m/z) [M + H+] calculated for C40H62NO4 620.4679. Found: 620.4676; HPLC analysis: peak area 98.0%, tR=9.32 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
6f: compound 6f was prepared using 1.1 equivalents of 3,4-dimethoxyphenylethylamine at room temperature for 72 h. Yield = 50%; Colorless oil; Rf (PhMe/EtOAc 9:1) = 0.10; 1H NMR (600 MHz, CDCl3) δ 6.81 (d, J = 8.1 Hz, 1 H), 6.74–6.69 (m, 2 H), 4.68 (d, J = 2.6 Hz, 1 H), 4.67–4.64 (m, 1 H), 4.58 (t, J = 1.7 Hz, 1 H), 4.26 (d, J = 12.4 Hz, 1 H), 3.87 (s, 3 H), 3.86 (s, 3 H), 3.84–3.81 (m, 1 H), 3.47–3.36 (m, 2 H), 3.18 (dd, J = 11.4, 4.7 Hz, 1 H), 2.76 (t, J = 6.9 Hz, 2 H), 2.44 (td, J = 10.7, 5.7 Hz, 1 H), 1.98 (qui, J = 9.1 Hz, 1 H), 1.81–1.69 (m, 3 H), 1.67 (s, 3 H), 1.65–1.48 (m, 7 H), 1.39 (m, 6 H), 1.30–1.18 (m, 5 H), 1.04 (s, 3 H), 1.02–0.99 (m, 1 H), 0.96 (s, 6 H), 0.89 (td, J = 12.9, 4.0 Hz, 1 H), 0.82 (s, 3 H), 0.76 (s, 3 H), 0.67 (d, J = 9.4 Hz, 1 H) 13C NMR (151 MHz, CDCl3) δ 158.4, 157.0, 149.0, 147.7, 120.7, 118.8, 112.0, 111.4, 109.8, 79.0, 55.9, 55.8, 55.3, 50.4, 48.8, 47.7, 46.6, 42.7, 42.3, 40.9, 38.9, 38.7, 37.5, 37.1, 35.7, 34.2, 29.6, 28.0, 27.4, 27.1, 25.2, 20.8, 19.1, 18.3, 16.1, 16.0, 15.3, 14.7; HR-ESI (m/z) [2M + H+] calculated for [C41H63NO5] 1299.9492. Found: 1299.9467. HPLC analysis: peak area 96.4%, tR=8.35 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
6g: compound 6g was prepared using 1.1 equivalents of 3,4,5-trimethoxyphenylethylamine at room temperature for 72 h. Yield = 55%; Colorless oil; Rf (PhMe/EtOAc 9:1) = 0.25; 1H NMR (600 MHz, CDCl3) δ 6.40 (s, 2 H), 4.71–4.69 (m, 1 H), 4.69 (d, J = 2.5 Hz, 1 H), 4.58 (t, J = 1.9 Hz, 1 H), 4.27 (d, J = 10.9 Hz, 1 H), 3.88–3.86 (m, 1 H), 3.85 (s, 6 H), 3.83 (s, 3 H), 3.44 (qui, J = 6.4 Hz, 2 H), 3.18 (dd, J = 11.4, 4.8 Hz, 1 H), 2.76 (t, J = 7.2 Hz, 2 H), 2.49–2.39 (m, 1 H), 1.99 (qui, J = 9.2 Hz, 1 H), 1.82–1.71 (m, 3 H), 1.68 (s, 3 H), 1.65–1.48 (m, 8 H), 1.39 (m, 6 H), 1.29–1.17 (m, 4 H), 1.04 (s, 3 H), 1.03-1.00 (m, 1 H), 0.97 (s, 6 H), 0.90 (td, J = 12.9, 4.0 Hz, 1 H), 0.82 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.4 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 157.0, 153.3, 150.2 137.9, 129.0, 128.2, 125.3, 109.8, 105.7, 79.0, 63.0, 60.8, 56.1, 55.3, 50.4, 48.8, 47.7, 46.6, 42.7, 42.2, 40.9, 38.8, 38.7, 37.5, 37.1, 36.6, 34.5, 34.2, 29.8, 29.6, 28.0, 27.4, 27.1, 25.2, 21.4, 20.8, 19.1, 18.3, 16.1, 16.0, 15.3, 14.8; HR-ESI (m/z) [M + H+] calculated for C42H66NO6 680.4890. Found: 680.4791; HPLC analysis: peak area 95.2%, tR=8.99 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
6h: compound 6h was prepared using 20 equivalents of piperazine at room temperature for 18 h. Yield = 75%; Colorless oil; Rf (DCE/MeOH 9:1) = 0.36; 1H NMR (600 MHz, CDCl3) δ 4.68 (d, J = 2.6 Hz, 1 H), 4.58 (t, J = 1.7 Hz, 1 H), 4.27 (d, J = 10.7 Hz, 1 H), 3.86 (d, J = 10.8 Hz, 1 H), 3.72 (s, 1 H), 3.51 (br s, 4 H), 3.17 (dd, J = 11.4, 4.8 Hz, 1 H), 2.89 (br s, 4 H), 2.80–2.62 (m, 2 H), 2.45 (td, J = 11.1, 5.0 Hz, 1 H), 2.03–1.93 (m, 1 H), 1.83–1.79 (m, 1 H), 1.78–1.69 (m, 3 H), 1.67 (s, 3 H), 1.66–1.48 (m, 5 H), 1.43–1.33 (m, 5 H), 1.31–1.15 (m, 4 H), 1.10–1.04 (m, 2 H), 1.03 (s, 3 H), 0.97 (s, 3 H), 0.96 (s, 3 H), 0.89 (td, J = 12.9, 4.2 Hz, 1 H), 0.81 (s, 3 H), 0.75 (s, 3 H), 0.67 (d, J = 9.3 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 155.8, 150.2, 129.0, 128.2, 109.8, 78.9, 63.9, 55.3, 50.4, 48.8, 47.7, 46.6, 45.3, 43.4, 42.7, 40.9, 38.8, 38.7, 37.5, 37.1, 34.6, 34.2, 30.0, 29.6, 28.0, 27.4, 27.1, 25.2, 20.8, 19.1, 18.3, 16.1, 16.1, 15.4, 14.8; HR-ESI (m/z) [M + H+] calculated for C35H59N2O3 555.4457. Found 555.4487; HPLC analysis: peak area 98.2%, tR=12.34 min, gradient: 45% (3 min to 45%) to 100% B over 20 min.
6i: compound 6i was prepared using 1.1 equivalents of N-methylpiperazine without DIPEA at room temperature for 15 h. Yield = 68%; White solid; m.p.: 189–190 oC; Rf (PhMe/EtOAc 1:9) = 0.13; 1H NMR (600 MHz, CDCl3) δ 4.69 (d, J = 2.5 Hz, 1 H), 4.59 (t, J = 1.7 Hz, 1 H), 4.29 (d, J = 9.3 Hz, 1 H), 3.88 (d, J = 10.9 Hz, 1 H), 3.68 (br s, 4 H), 3.18 (dd, J = 11.6, 4.8 Hz, 1 H), 2.78–2.48 (m, 4 H), 2.47–2.39 (m, 1 H), 2.02–1.94 (m, 1 H), 1.83–1.69 (m, 4 H), 1.68 (s, 3 H), 1.66–1.47 (m, 6 H), 1.43–1.36 (m, 5 H), 1.30–1.15 (m, 7 H), 1.11–1.05 (m, 2 H), 1.04 (s, 3 H), 0.98 (s, 3 H), 0.97 (s, 3 H), 0.93 − 0.85 (m, 2 H), 0.82 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.6 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 155.6, 150.1, 129.0, 128.2, 125.3, 109.9, 91.5, 79.0, 64.2, 55.3, 54.2, 50.4, 48.8, 47.7, 46.6, 42.7, 40.9, 38.9, 38.7, 37.5, 37.1, 34.6, 34.2, 30.0, 29.6, 28.0, 27.4, 27.1, 25.2, 20.8, 19.1, 18.3, 16.1, 16.1, 15.3, 14.8; HR-ESI (m/z) [M + H+] calculated for C36H61N2O3 569.4682. Found: 569.4669; HPLC analysis: peak area 98.0%, tR=9.65 min, gradient: 25% (3 min to 25%) to 75% B over 20 min.
6j: compound 6j was prepared using 1.1 equivalents of tert-butyl-4-(2-aminoethyl) piperazine-1-carboxylate at room temperature for 20 h. Yield = 70%; Colorless oil; Rf (EtOAc) = 0.4; 1H NMR (600 MHz, CDCl3) δ 5.08 (s, 1 H), 4.68 (d, J = 2.6 Hz, 1 H), 4.58 (t, J = 2.0 Hz, 1 H), 4.30–4.24 (m, 2 H), 3.85 (d, J = 11.1 Hz, 1 H), 3.71 (td, J = 5.8, 1.7 Hz, 2 H), 3.42 (br s, 4 H), 3.30 (br s, 1 H), 3.18 (dd, J = 11.6, 4.8 Hz, 1 H), 2.50–2.47 (m, 1 H), 2.39 (br s, 4 H), 2.02–1.95 (m, 1 H), 1.85 (d, J = 12.9 Hz, 1 H), 1.79 (dd, J = 12.4, 7.3 Hz, 1 H), 1.74 (d, J = 12.5 Hz, 1 H), 1.68 (s, 3 H), 1.65–1.48 (m, 9 H), 1.46 (s, 9 H), 1.43–1.35 (m, 5 H), 1.31–1.22 (m, 5 H), 1.04 (s, 3 H), 0.97 (s, 3 H), 0.96 (s, 3 H), 0.92 − 0.85 (m, 1 H), 0.82 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.3 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 171.1, 157.1, 154.7, 150.3, 129.0, 128.2, 125.3, 109.8, 79.7, 79.0, 64.3, 63.1, 57.2, 55.3, 52.7, 50.4, 48.8, 47.7, 46.6, 42.7, 40.9, 38.9, 38.7, 37.5, 37.1, 34.6, 34.2, 30.6, 29.7, 28.4, 28.0, 27.4, 27.1, 25.2, 20.8, 19.1, 18.3, 16.1, 16.0, 15.3, 14.8, 13.7; HR-ESI (m/z) [M + H+] calculated for C42H72N3O5 698.5472. Found: 698.5447; HPLC analysis: peak area 98.7%, tR=8.27 min, gradient: 25% (3 min to 25%) to 75% B over 20 min.
6l: compound 6l was prepared using 1.1 equivalents of 4-(4-Methylpiperazino) benzylamine at room temperature for 24 h. Yield = 42%; White solid; m.p.: 151–152 oC; Rf (EtOAc/Et3N 10:0.1) = 0.33; 1H NMR (600 MHz, CDCl3) δ 7.21–7.17 (m, 2 H), 6.90 (s, 1 H), 6.88 (s, 1 H), 4.88 (br s, 1 H), 4.68 (d, J = 2.5 Hz, 1 H), 4.58 (t, J = 1.9 Hz, 1 H), 4.30–4.24 (m, 3 H), 3.87 (d, J = 11.2 Hz, 1 H), 3.29 (br s, 4 H), 3.18 (d, J = 11.1 Hz, 1 H), 2.72 (br s, 4 H), 2.48–2.41 (m, 4 H), 2.01–1.94 (m, 1 H), 1.84–1.72 (m, 3 H), 1.67 (s, 3 H), 1.63–1.48 (m, 6 H), 1.44–1.34 (m, 5 H), 1.29–1.17 (m, 6 H), 1.04 (s, 3 H), 1.02–0.99 (m, 1 H), 0.96 (s, 6 H), 0.89 (td, J = 13.1, 4.4 Hz, 2 H), 0.82 (s, 3 H), 0.76 (s, 3 H), 0.67 (d, J = 9.5 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 171.1, 157.0, 150.3, 129.0, 128.8, 128.2, 116.4, 109.7, 79.0, 63.2, 60.4, 55.3, 54.7, 50.4, 48.8, 48.6, 47.7, 46.6, 44.6, 42.7, 40.9, 38.9, 38.7, 37.5, 37.1, 34.5, 34.2, 29.8, 29.7, 28.0, 27.4, 27.1, 25.2, 21.0, 20.8, 19.1, 18.3, 16.1, 16.1, 15.3, 14.7, 14.2; HR-ESI (m/z) [M + H+] calculated for C43H68N3O3 674.5260. Found: 674.5258; HPLC analysis: peak area 96.4%, tR=11.5 min, gradient: 50% (3 min to 50%) to 100% B over 20 min.
6m: compound 6m was prepared using 1.1 equivalents of 6,7-dimethoxy-1,2,3,4-tetrahydro-isoquinoline at room temperature for 17h. Yield = 42%; Rf (PhMe/EtOAc9:1) = 0.18; Colorless oil; 1H NMR (600 MHz, CDCl3) δ 6.62 (s, 1 H), 6.60 (br s, 1H), 4.69 (d, J = 2.6 Hz, 1 H), 4.59 (t, J = 1.9 Hz, 1 H), 4.55 (d, J = 10.3 Hz, 2 H), 4.31 (d, J = 10.4 Hz, 1 H), 3.90 (d, J = 10.9 Hz, 1 H), 3.86 (s, 6 H), 3.72–3.63 (m, 2 H), 3.18 (dd, J = 11.5, 4.7 Hz, 1 H), 2.77 (s, 2 H), 2.48 (td, J = 11.2, 5.8 Hz, 1 H), 2.06–1.96 (m, 1 H), 1.93–1.81 (m, 2 H), 1.78–1.70 (m, 2 H), 1.69 (s, 3 H), 1.67–1.48 (m, 8 H), 1.45–1.35 (m, 5 H), 1.31–1.17 (m, 4 H), 1.07–1.05 (m, 1 H), 1.04 (s, 3 H), 0.99 (s, 3 H), 0.97 (s, 3 H), 0.90 (td, J = 13.5, 4.7 Hz, 1 H), 0.82 (s, 3 H), 0.76 (s, 3 H), 0.68 (d, J = 9.4 Hz, 1 H); 13C NMR (151 MHz, CDCl3) δ 150.2, 147.7, 147.7, 137.9, 129.0, 128.2, 125.3, 111.5, 109.8, 79.0, 63.8, 56.0, 56.0, 55.3, 50.4, 48.8, 47.7, 46.7, 42.7, 40.9, 38.9, 38.7, 37.5, 37.2, 34.7, 34.2, 30.1, 29.7, 28.0, 27.4, 27.2, 25.2, 21.4, 20.8, 19.2, 18.3, 16.1, 15.3, 14.8; HR-ESI (m/z) [M + H+] calculated for C42H64NO5 662.4784. Found: 662.4775; HPLC analysis: peak area 98.6%, tR=10.90 min, gradient: 90% (3 min to 90%) to 95% B over 5 min and from 95–100% B over 10 min.
Deprotection of compound 6j
To an ice-cold solution of 6j (9 mg, 0.01 mmol) in DCM (40 µL), trifluoroethanol (0.05mmol, 5µL) and TFA (0.25mmol, 20µL) were added and the reaction mixture was stirred at ambient temperature for 3 h. Volatile components were evaporated under vacuum, the oily residue was triturated with a mixture of diethyl ether (Et2O)/ hexane, and refrigerated overnight. The desired trifluoroacetate salt 6k was afforded as pale-yellow oil. Yield = 90%; HR-ESI (m/z) [M+] calculated for C37H65N3O3+ 598.4942. Found: 598.4932; HPLC analysis: peak area 98.4%, tR=8.28 min, gradient: 25% (3 min to 25%) to 75% B over 20 min.
Material for biological studies. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), histopaque 1077, lectin from Phaseolus vulgaris (PHA), and Rho123 were obtained from Sigma Aldrich (Sigma-Aldrich Co., St Louis, MO). Doxorubicin hydrochloride (Dox, 99.8%, Synbias Pharma Ltd.) was obtained from Nanox Release Technology (Buenos Aires, Argentina) and was prepared immediately prior to use dissolved in bi-distilled water. Verapamil hydrochloride 98.0% was provided by Parafarm (Buenos Aires, Argentina) while tariquidar was purchased from ChemFaces Biochemical Co., Ltd (Wuhan, China). Both were prepared in DMSO. Sterile plastic material was purchased from Greiner Bio-One (Frickenhausen, Germany). Flow cytometry was carried out in a Life Technologies Attune-NxT flow cytometer with 96-well autosampler (Thermo Fisher Scientific, USA).
Cell lines and cell culture. The sensitive CML cells K562 and its counterpart P-gp-overexpressing Lucena 1 cells, were cultured in RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin (Invitrogen Life Technologies, Carlsbad, CA) at 37°C in a 5% CO2 humidified atmosphere. Both cell lines were provided by Dr. V. Rumjanek, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil 44. Passages were performed twice a week and cells in exponential growth phase with a viability of 90%, determined by trypan blue exclusion, were used for the experiments. To give rise to the MDR phenotype, resistant lines were subjected to continuous treatment with 60 nM of Dox. This treatment was interrupted 4 days before each trial. Lucena 1 displayed 32.3-fold more resistance to Dox than K562, as evidenced by the IC50 values obtained by MTT assay (IC50 = 84.21 ± 7.08 and 2.61 ± 0.70 µM, respectively). This level of resistance correlates with the level of expression of P-gp 26.
MTT colorimetric assay. The cytotoxic effect of the compounds and of the gold standards verapamil and tariquidar was determined against Lucena 1 and K562 using MTT assay. This colorimetric technique is based on the ability of viable cells to capture MTT and reduce it through the mitochondrial enzyme succinic dehydrogenase to its insoluble form, formazan crystals 45. The target compounds dissolved in DMSO (0.5% v/v as final concentration which was non-toxic to cells) at a maximum concentration of 50 µM were exposed to 5 x 104 cells distributed in a 96-well plate and incubated for 48 h. At the end of the incubation period, the cells were treated with 5 mg/mL of MTT solution prepared in phosphate buffered saline (PBS) and incubated at 37°C in a humid atmosphere with 5% CO2 for another 4 h. After this time, the plate was centrifuged at 2000 rpm for 10 min, and 100 µL of DMSO was added to dissolve the formazan crystals formed. Absorbance (Abs) was read at a wavelength of 595 nm in an iMark microplate reader (Bio-Rad, USA). The percentages of cytotoxicity were determined as compared to DMSO treated control cells, which were considered 100% viable as showing the same level of growth as the untreated cells. Those compounds showing a cytotoxicity > 40% at 50 µM were further assayed with a range of two-fold decreasing concentrations for their IC50 values, which were calculated as previously reported 46,47 using non-linear regression by GraphPad Prism 7 (GraphPad Software, Inc., San Diego, CA, USA, www.graphpad.com).
Rhodamine 123 and doxorubicin intracellular accumulation assays. The ability of the compounds to increase the intracellular concentration of Rho123 or of Dox by the inhibition of P-gp was evaluated using flow cytometry, as described earlier 27,28,48. Lucena 1 or K562 cells adjusted to a density of 5x104 cells/well were seeded in 96-well plates, previously containing RPMI-1640 medium in the presence of each compound dissolved in DMSO, at concentrations ranging from 0.049 to 50 µM and from 6.25 to 50 µM for each cell line, respectively. Cells treated only with DMSO at 0.5% v/v were used as negative controls, while those treated with the standard P-gp inhibitors 33, verapamil and tariquidar were used as positive controls. The samples were incubated for 1 h at 37°C in a humid atmosphere with 5% CO2, after which the corresponding fluorescent substrate, Rho123 (500 ng/mL) or Dox (5 µM), was added, and a second incubation was carried out under the same conditions for 1 h. Subsequently, cells were thoroughly washed twice with cold PBS and kept in dark and cold. Samples were gated on a forward versus size scatter, and 15,000 events from each were analyzed to determine the cell-associated fluorescence. It is important to notice that the target derivatives were not fluorescent by themselves (data not shown).
Dox and Rho123 were excited at 488 nm, and the fluorescence emitted was collected with 585/42 nm or 530/30 nm bandpass filters, respectively. Mean fluorescence intensity (MFI) was analyzed using Flowjo software (Tree Star, Inc. Ashland, OR). The blocking of the P-gp transport function was evaluated by the parameter fluorescence intensity ratio (FIR), calculated as the ratio of the MFI of Rho123 or Dox with the addition of the likely inhibitor to the MFI of Rho123 or Dox alone.
Doxorubicin resistance reversal assay. These experiments were undertaken to evaluate the ability of the two most effective compounds to potentiate Dox cytotoxicity by inhibiting the outward transport mediated by P-gp. MTT staining was used, for which 5 x 104 Lucena 1 and K562 cells/well and increasing concentrations of Dox (3.4–431 and 0.33–34.5 µM, respectively) in the absence or presence of 0.024 to 1.56 µM and 0.19 to 1.56 µM of compounds 6g and 6i, respectively, were incubated in 96-well plates containing RPMI-1640 medium. DMSO (0.5% v/v) was used as a negative control, while verapamil was chosen as standard inhibitor. Simultaneously, viability controls were run with no addition of the dissolution solvents. The cells were incubated at 37°C at 5% CO2 for 48 h and the same protocol described above was followed. The potency of the compounds was established by means of the fold reversal (FR) values calculated from dividing the IC50 of Dox alone by the IC50 of Dox with different concentrations of each compound tested.
Cytotoxicity on peripheral blood mononuclear cells and hemolysis assay. In order to preliminarily determine the safety of the active compounds 6g and 6i in normal cells, different concentrations (1.56-50 µM) of these were assayed by the MTT technique on peripheral blood mononuclear cells (PBMC) according to published works 28,48. PBMC were isolated by the Ficoll-Hypaque density gradient centrifugation technique of fresh heparinized blood from healthy human volunteers. The protocol was previously approved by the Catholic University of Córdoba Research Ethics Board and signed informed consents were obtained from donors. All methods were performed in accordance with the relevant guidelines and regulations. Briefly, 1x105 cells/well in a 96-well plate were incubated with 10 µg/mL PHA, in the presence of the corresponding concentrations of the compounds or 0.5% v/v DMSO (negative control) for 48 h, and processed as above described for the MTT method. The percentages of inhibition on cell proliferation were determined by comparison with negative controls, and the corresponding IC50 values were calculated from the best fit of the concentration-dependent inhibition curves in GraphPad Prism 7.
For the erythrocyte hemolysis assay, the red blood cell suspension was obtained from fresh heparinized blood collected from healthy human volunteer donors, without receiving any treatment. The blood sample was centrifuged at 2,500 rpm for 10 minutes at 4°C. The supernatant was discarded, and the red blood cell pellet was subjected to two consecutive washes with sterile PBS, using the same centrifugation conditions. An aliquot of the pellet obtained was resuspended in PBS (3% v/v). The compounds were incubated at decreasing concentrations starting from 50 µM with the suspension at 37°C for 1 h. Controls with 1% DMSO and no solvent were included.
Statistical analysis. The results are expressed as the mean ± standard error (SE). Data were analyzed using the one-tailed paired Student's t-test and one-tailed unpaired Student's t-test, as appropriate, by GraphPad Prism 7.0. p values ≤ 0.05 were considered statistically significant. Each experiment was performed in triplicate or quadruplicate and done independently at least three times.
Molecular Modeling. The structural model of the P-gp was based on the structure cocrystallized with paclitaxel (PDB entry 6QEX) 41, and prepared as described in detail in Laiolo et al. 28. In this work, the structure 7A69, cocrystallized with the antitumoral agent vincristine 42, was also used for checking the docking procedure, although the simulations of vincristine were made on the same receptor (based on 6QEX) used for the rest of the substrates and inhibitors.
The docking protocol was fairly similar to that in Laiolo et al. 28. Briefly, the box included the whole TM region (no assumption about a particular binding site) and the docking was run with Autodock Vina 1.1.2-5 49, collecting the first 12 lowest energy poses or those within 3 kcal/mol above the lowest. The “exhaustiveness” parameter was set between 64 and 192 (much bigger than the default, 8, due to the size of the region), and repeating the simulation at least 12 times. Standard Gagsteiger’s charges 49 were set for all ligands except for Rho123, for which RESP 50 at the CAM-B3LYP/6–31 + G(d) 51 charges were used to correctly draw the charge delocalization of the cationic dye. The structure of the ligands were obtained by full geometry optimization with the semiempirical PM6 Hamiltonian as implemented in Gaussian 16 Rev. A03 52 The structures were confirmed as minima by diagonalization of the Hessian matrix.
The initial structures for the MD simulations were prepared from the most stable docked structures using the AMBER18 53 utilities LEaP, parmchk2 and antechamber 54. The atomic charges on the ligands were obtained with the AMBER18 module sqm, using AM1 quantum calculations and the parameters assigned based on the auxiliary force field GAFF. 53. The protocol for running the MD simulations was successfully applied in Laiolo et al. 27,28. This involves: I) 250 steepest descent minimization steps of the whole system, keeping the protein tightly restrained and embedded into a box of TIP3P water molecules with a minimum distance of 10 Å to each wall, and enough chloride counter-ions to reach electro-neutrality; II) 6500 conjugate gradient minimization steps of the whole system; III) 100 ps slowly heating in the NTV ensemble with the protein positionally restrained in the backbone; IV) 60 ns of simulation in the NTP ensemble, at 1 atm and 300 K. Procedures III-IV were repeated in two or three independent trajectories using the Andersen thermostat and barostat 55. In the reasonably equilibrated system, the RMSd of the ligand and all residues surrounding it within 5Å was in most cases between 1.1 and 1.6 Å, excluding hydrogens (examples in Supplementary Material Fig. S6). Due to the transmembrane nature of P-gp, a 50.0 kcal/Å2 harmonic restraint was kept for the backbone atoms. Electrostatic interactions were computed using the Particle Mesh Ewald (PME) method with a cutoff of 10 Å 56. The SHAKE algorithm, as implemented in pmemd.cuda, was applied to constrain hydrogen-heavy atom bonds 53. The force field used for the atoms of the protein was ff14SB 57.The trajectory analyses were made with cpptraj 53 and VMD 1.9.3 58, with the latter also used for graphics rendering.
The free energy calculations were made using the mmpbsa module of AMBER 18 by applying Poisson-Boltzman (PB) and Generalized Born (GB) models 59. The energy analyses were made for the last 8–12 ns of simulation as the average over at least two independent trajectories. The frames were sampled once each 10–20 ps in order to keep the energy self-correlation negligible. A detailed analysis of the dynamics of the contacts was made for all simulated compounds by partitioning the energy contribution of each amino acid with the inhibitor from one representative run as shown for Rho123, vincristine, and compounds 6g, 6i, 6f, 3a, and 3d displayed in Fig. 8. For comparison purposes, the pattern obtained for compound 6m (which slightly departs in its mode of binding from the subset of compounds that behave as compound 6i) is shown in the Supplementary Material Fig. S5, together with data previously obtained using the same modeling models for doxorubicin and tariquidar as other reference agents run as substrate and inhibitor, respectively 28.
The average total number of H-bonds were in general between 0.5 and 1.5 during the equilibrated trajectories, with some examples shown as Supplementary Material Fig. S7. Chimera 1.15 60 was used for clustering analyses, using the last 50 ns of each trajectory and for the graphics rendering of representative conformations thus obtained.
{kind=link}
{kind=link}