Chemistry
General procedures
All reagents were purchased from commercial suppliers and used without further purification. Standard HPLC-grade quality solvents were used. Anhydrous THF, DCM, and DMF were obtained from a Glass Contour Solvent System (SG Water USA). Anhydrous MeOH was obtained by distillation over CaH2 and stored over activated 3 Å molecular sieves for a minimum of 24 h (according to standard protocols). Et3N and pyridine were kept dry by storage over activated 3 Å molecular sieves. Thin-layer chromatography (TLC) were performed using Merck aluminum sheets covered with silica gel C-60 F254. Visualization of TLC plates was performed using UV light (254 nm), KMnO4 stain, or p-anisaldehyde stain prepared according to standard procedures. Flash column chromatography was performed using glass columns packed with Merck Geduran Si 60 (0.040 − 0.063 mm) as a stationary phase, or with a Combi Flash NextGen 300 + system (Teledyne) on RediSep Gold® silica gel flash columns of the appropriate size. Eluent systems are reported as volume ratios, and Rf value are specified under each protocol. 1D and 2D NMR spectra were acquired using a Bruker Avance II equipped with a 5 mm broad band probe (BBFO) operating at 400 MHz for 1H NMR and 101 MHz for 13C NMR or a Bruker Avance III HD equipped with a cryogenically cooled 5 mm dual probe optimized for 13C and 1H NMR operating at 600 MHz for 1H NMR and 151 MHz for 13C NMR. HSQC, HMBC, and ROESY experiments were used to support analyses when 1H NMR, 13C NMR, and COSY were inadequate. Chemical shifts (δ) are reported in ppm downfield from TMS (δ = 0) using either the solvent resonance as the internal standard (CDCl3, 1H: 7.26 ppm, 13C: 77.16 ppm; DMSO-d6, 1H: 2.50 ppm, 13C: 39.52 ppm; MeOD, 1H: 3.31 ppm, 13C: 49.00 ppm; D2O, 1H: 4.79 ppm) or 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt (TMSP, 13C: 0 ppm). Coupling constants (J) are reported in Hz, and the field is reported in each case. Multiplicities are reported as singlet (s), broad singlet (bs), doublet (d), doublet of doublets (dd), doublet of triplets (dt), doublet of doublet of doublets (ddd), doublet of doublet of triplets (ddt), triplet (t), triplet of doublets (td), quartet (q), pentet (p), septet (sep), and multiplet (m). Mass spectrometric data was recorded using either a LC-MS system built from an Agilent 1200 series solvent delivery system equipped with an autoinjector coupled to a DAD and an Agilent 6130A series quadrupole electrospray ionization detector or a Waters Aquity UPLC-MS equipped with a dual-wavelength PDA (214 and 254 nm) combined with electrospray ionization. Gradients of H2O/MeCN/HCOOH (95:5:0.1) (solvent A) and MeCN/HCOOH (100:0.1) (solvent B) were employed. Purity was assessed by analytical HPLC on an UltiMate HPLC system (Thermo Scientific) consisting of an LPG-3400A pump (1 mL/min), a WPS-3000SL autosampler, and a DAD-3000D diode array detector using a Gemini-NX C18 column (4.6 × 250 mm, 3 µm, 110 Å); gradient elution was 0 to 100% B (MeCN/H2O/TFA, 90:10:0.1) in solvent A (H2O/TFA, 100:0.1) over 20 min. Data were acquired and processed using Chromeleon Software v. 6.80. For final compounds (12–20, 22, 23) analytical purity is ≥ 95%; retention times (tR) are indicated. Chiral analytical HPLC was performed using an Hitachi 7000 series HPLC apparatus consisting of a D-7000 interface unit, an L-7100 pump (0.8 mL/min), a L-7200 autosampler, a L-7400 UV variable detector (detection at 190 nm), using a Astec Chirobiotic T column (5 µm, 4.6 × 250 mm); isocratic elution with 60% solvent A (milliQ water) and 40% solvent B (methanol) was performed. Enantiomeric excesses (ee) of compounds (S,S)-6a and (S,S)-7a were higher than 99% and retention times (Rt) are reported. Preparative HPLC purification was carried out on a Dionex Ultimate 3000 HLPC system consisting of an LPG-3200BX pump (20 mL/min), a Rheodyne 9725i injector, a 10 mL loop, an MWD-3000SD detector (200, 210, 254, and 281 nm), and an AFC-3000SD automated fraction collector using a Gemini-NX C18 column (21.2 × 250 mm, 5 µm, 110 Å). Solvent A (H2O/TFA, 100:0.1) and solvent B (MeCN/H2O/TFA, 90:10:0.1) were utilized. Unless specified otherwise, a gradient elution from 0% B to 100% B was used, over 20 min. Data were acquired and processed using Chromeleon Software v.6.80. Mosher analysis of intermediates (S)-27a and (S)-28a was performed according to protocols reported in literature.
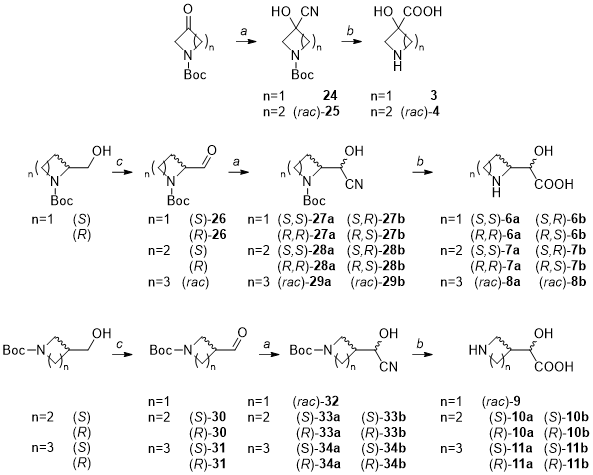
METHOD A: general procedure for the preparation of compounds 24, 25, 27–29, 32–34, 37.
The appropriate aldehyde or ketone (5.02 mmol, 1 eq) was dissolved in diethyl ether (5 mL). A solution of NaHSO3 (1.2 eq) in deionized water (10 mL) was added. After stirring for 15 minutes, KCN (1.5 eq) was carefully added, and the reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with diethyl ether (30 mL), the phases separated, and the water layer was extracted with diethyl ether (3 × 10 mL). The combined organic phases were dried over anhydrous sodium sulfate, filtered, and the solvent was evaporated under reduced pressure. The crude was purified by silica gel flash column chromatography as specified, providing the desired compounds 24, 25, 27–29, 32–34 and 37 in variable yields (11–90%).
METHOD B: general procedure for the preparation of compounds 3, 4, 6–11, 21.
The α-hydroxynitrile of interest (0.40 mmol) was suspended in HCl 4M (1 mL) and heated to 85 °C overnight. Upon completion, the solution was cooled to rt, the volatiles were removed under reduced pressure and the residue was desalted by ion exchange chromatography (5M NH3aq). The crude purified as specified, providing the desired products 3, 4, 6–11, 21 as off-white solids in variable yields (17–75%).
METHOD C: general procedure for the preparation of compounds 26, 30, 31.
The appropriate primary alcohol (25.8 mmol, 1 eq) was dissolved in DCM (50 mL), and trichloroisocyanuric acid (1.05 eq) was added portion-wise. Upon cooling to 0 °C, TEMPO (0.01 eq) was slowly added. The reaction was allowed to warm at rt and stirred for 15 minutes at rt. Upon completion, the mixture was filtered over celite®, and the resulting solution was washed with a saturated solution of Na2CO3 (3 × 15 mL), HCl 1M (3 × 15 mL), and brine (10 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, the solvents were removed under reduced pressure. The crude was purified by silica gel flash column chromatography when specified, providing the desired products 26, 30 and 31 in moderate to excellent yields (42–95%).
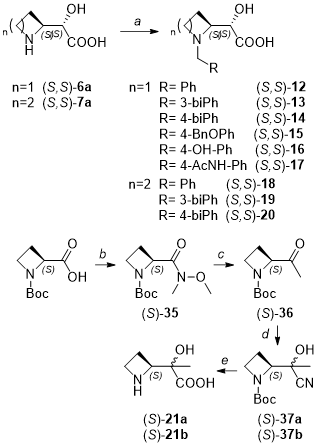
METHOD D: general procedure for the preparation of compounds (S,S)-12-20.
In an oven-dried tube under inert atmosphere, intermediate (S,S)-6a or (S,S)-7a (0.31 mmol, 1 eq), the appropriate aromatic aldehyde (1 eq) and NaBH3CN (1 eq) were suspended in dry methanol (2 mL). The reaction mixture was stirred at 60 °C overnight. Upon cooling, the mixture was quenched with 2 mL water and diluted with methanol until a clear solution was formed. Purification by preparative HPLC provided the desired compounds (S,S)-12-20 in moderate to good yields (31–89%).
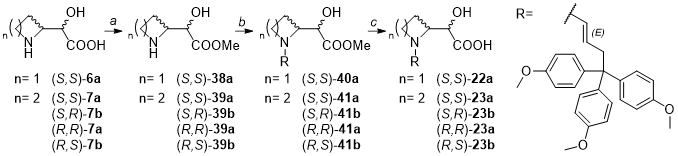
METHOD E: general procedure for the preparation of compounds 38 and 39.
To an ice-cooled stirred solution of the appropriate carboxylic acid (S,S)-6a or (S,S)-7a (0.90 mmol, 1 eq) in methanol (2.5 mL), thionyl chloride (4 eq) was carefully added dropwise over 5 min, and the reaction mixture was left stirring at rt overnight. The volatiles were evaporated under reduced pressure, and the residue was diluted with EtOAc (3 mL), cooled to °0 C, and water (3 mL) was added. The pH of the water layer was adjusted to 10 by dropwise adding a 4M solution of NaOH. The two phases were separated and water layer was extracted with EtOAc (3 x 10 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and the solvents were evaporated under reduced pressure providing the desired compounds 38 and 39 in very good yields (75–92%).
METHOD F: general procedure for the preparation of compounds 40 and 41.
To a flame-dried two-neck flask under inert atmosphere, the appropriate diasteromer of intermediate 38 or 39 (0.32 mmol, 1 eq) was dissolved in 3 mL of acetone/DMF/ACN 1:1:1. K2CO3 (3 eq) and KI (0.1 eq) were added, and the suspension was stirred for 30 min. Afterwards, (E)-4,4',4''-(4-bromobut-2-ene-1,1,1-triyl)tris(methoxybenzene) 42 (1.3 eq) was added, and the reaction mixture was stirred at rt overnight in the dark. Upon completion, 10 mL of EtOAc were added, the resulting suspension was filtered and the filtrate was evaporated under reduced pressure, providing a crude that was purified by preparative HPLC. The desired compounds 40 and 41 were obtained in low yields (14–37%).
METHOD G: general procedure for the preparation of compounds 22 and 23.
To the appropriate methyl ester 40 or 41 (0.07 mmol, 1 eq), a 1:1 mixture of LiOH 2M in water/dioxane (1 mL) was added. The reaction mixture was stirred for 2 hours at rt and then dioxane was evaporated under reduced pressure. The resulting crude was purified by preparative HPLC, providing the desired compounds 22 and 23 in moderate yields (32–74%).
Synthesis of tert-butyl 3-cyano-3-hydroxyazetidine-1-carboxylate ( 24 ).
Obtained from of tert-butyl 3-oxoazetidine-1-carboxylate (500 mg, 2.90 mmol), according to METHOD A as a white solid in 90% yield. 1H NMR (400 MHz, CDCl3) δ 4.38 (d, J = 9.6 Hz, 2H), 4.08 (d, J = 9.6 Hz, 2H), 1.45 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 156.0, 119.2, 81.5, 60.6, 28.4. Rf (1:1 heptane/EtOAc) = 0.50 Rt (LC-MS) = 3.651 min. LC-MS (ESI): m/z calcd. for C5H7N2O3 [M + H-tBu]+= 143.12, found 143.1.
Synthesis of 3-carboxy-3-hydroxyazetidine hydrochloride ( 3 ).
Obtained from tert-butyl 3-cyano-3-hydroxyazetidine-1-carboxylate 24 (100 mg, 0.50 mmol), according to METHOD B. The residue was taken in HCl 1M, dried, and the solid was crystallized from isopropanol in 21% yield. Rt (LC-MS) = 0.492 min. LC-MS (ESI): m/z calcd. for C4H7NO3 [M + H]+= 118.05, found 118.0. 1H NMR (600 MHz, MeOD) δ 4.43 (d, J = 11.9 Hz, 2H), 4.08 (d, J = 11.9 Hz, 2H). 13C NMR (151 MHz, MeOD) δ 173.6, 71.9, 58.7.
Synthesis of (rac)-tert-butyl 3-cyano-3-hydroxypyrrolidine-1-carboxylate ((rac)- 25 ).
Obtained from of tert-butyl 3-oxopyrrolidine-1-carboxylate (1.00 g, 5.4 mmol), according to METHOD A as a white solid in 61% yield. Rf (1:1 n-heptane/EtOAc) = 0.63. Rt (LC-MS) = 3.597 min. LC-MS (ESI): m/z calcd. for C6H9N2O3 [M + H-tBu]+= 157.06, found 157.1. 1H NMR (600 MHz, CDCl3) δ 4.02 (bs, 1H), 3.80–3.61 (m, 3H), 3.61–3.51 (m, 1H), 2.37–2.30 (m, 2H), 1.47 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 154.6, 154.3, 119.6, 81.0, 80.9, 70.8, 69.9, 57.8, 57.5, 44.0, 43.6, 38.7, 38.4, 28.6.
Synthesis of (rac)-3-hydroxypyrrolidine-3-carboxylic acid ((rac)- 4 ).
Obtained from (rac)-tert-butyl 3-cyano-3-hydroxypyrrolidine-1-carboxylate (rac)-25. (200 mg, 0.94 mmol), according to METHOD B. The crude was dissolved in water, precipitated with acetone at 0 °C and filtered, providing the desired compound as a off-white solid in 75% yield. Rt (LC-MS) = 0.474 min. LC-MS (ESI): m/z calcd. for C5H10NO3 [M + H]+= 132.07, found 132.1. 1H NMR (600 MHz, D2O) δ 3.65–3.59 (m, 2H), 3.54 (ddd, J = 11.7, 9.6, 7.5 Hz, 1H), 3.39 (dd, J = 12.3, 1.7 Hz, 1H), 2.42 (dt, J = 13.8, 9.6 Hz, 1H), 2.19 (dddd, J = 13.8, 7.5, 3.6, 1.7 Hz, 1H). 13C NMR (151 MHz, D2O) δ 180.5, 83.5, 58.2, 47.6, 39.5.
Synthesis of tert-butyl (S)-2-formylazetidine-1-carboxylate ((S)- 26 ).
Obtained from tert-butyl (S)-2-(hydroxymethyl)azetidine-1-carboxylate (900 mg, 4.81 mmol), according to METHOD C, as an oil in 48% yield after silica gel flash column chromatography (gradient from n-heptane to n-heptane/THF 1:1). Rf (2:1 n-heptane/THF) = 0.29. Rt (LC-MS) = 4.617 min. LC-MS (ESI): m/z calcd. for C18H31N2O6 [2M + H]+= 371.22, found 371.2. 1H NMR (400 MHz, CDCl3) δ 9.79 (s, 1H), 4.59 (t, J = 7.6 Hz, 1H), 4.03–3.87 (m, 2H), 2.52–2.37 (m, 1H), 2.36–2.19 (m, 1H), 1.44 (s, 9H).
Synthesis of tert-butyl (R)-2-formylazetidine-1-carboxylate ((R)- 26 ).
Obtained from tert-butyl (R)-2-(hydroxymethyl)azetidine-1-carboxylate (1.00 g, 5.30 mmol), according to METHOD C, as an oil in 42% yield after silica gel flash column chromatography (gradient from n-heptane to n-heptane/THF 1:1). TLC, LC-MS, 1H NMR data as for (S)-26.
Synthesis of tert-butyl (S)-2-((S)-cyano(hydroxy)methyl)azetidine-1-carboxylate (S,S)- 27a and of (S)-2-((R)-cyano(hydroxy)methyl)azetidine-1-carboxylate (S,R)- 27b .
Obtained from tert-butyl (S)-2-formylazetidine-1-carboxylate (S)-26 (570 mg, 3.08 mmol), according to METHOD A. Purification by silica gel flash column chromatography (gradient from DCM to DCM/ethyl acetate 9:1) allowed separation of the two diastereoisomers (S)-27a and (S)-27b. Diastereoisomer (S)-27a was obtained as a white solid in 48% yield. Rf (DCM/ethyl acetate 95:5) = 0.39. Rt (LC-MS) = 3.679 min. LC-MS (ESI): m/z calcd. for C6H9N2O3 [M + H-tBu]+= 157.06, found 157.1. 1H NMR (400 MHz, CDCl3) δ 6.25 (d, J = 10.5 Hz, 1H), 4.72–4.54 (m, 1H), 4.48 (dd, J = 10.5, 2.2 Hz, 1H), 4.04–3.84 (m, 2H), 2.43–2.26 (m, 1H), 2.25–2.08 (m, 1H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 158.2, 117.6, 82.2, 66.7, 63.9, 47.3, 28.4, 18.2. Mosher analysis and 1H NMR/conformational analysis determined (S,S) configuration for (S)-27a. Diastereoisomer (S)-27b was obtained as a white solid in 33% yield. Rf (DCM/ethyl acetate 95:5) = 0.21. Rt (LC-MS) = 3.523 min. LC-MS (ESI): m/z calcd. for C6H9N2O3 [M + H-tBu]+= 157.06, found 157.1. 1H NMR (400 MHz, CDCl3) δ 5.50 (bs, 1H), 4.64 (d, J = 8.3 Hz, 1H), 4.53 (td, J = 8.3, 6.6 Hz, 1H), 3.93 (td, J = 8.7, 7.2 Hz, 1H), 3.84 (td, J = 8.7, 4.7 Hz, 1H), 2.40 (dtd, J = 12.0, 8.7, 4.7 Hz, 1H), 2.21–2.06 (m, 1H), 1.46 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 158.0, 117.0, 82.2, 66.5, 64.0, 46.9, 28.4, 18.5. 1H NMR/conformational analysis determined (S,R) absolute configuration for (S)-27b.
Synthesis of tert-butyl (R)-2-((R)-cyano(hydroxy)methyl)azetidine-1-carboxylate (R,R)- 27a and of (R)-2-((S)-cyano(hydroxy)methyl)azetidine-1-carboxylate (R,S)- 27b.
Obtained from tert-butyl (R)-2-formylazetidine-1-carboxylate (R)-26 (340 mg, 1.84 mmol), according to METHOD A. Purification by silica gel flash column chromatography (gradient from DCM to DCM/EtOAc 9:1) allowed separation of the two diastereoisomers (R)-27a and (R)-27b. Diastereoisomer (R)-27a was obtained as a white solid in 42% yield. TLC, LC-MS, 1H NMR and 13C NMR data as for (S,S)-27a, confirming (R,R) configuration for (R)-27a. Diastereoisomer (R)-27b was obtained as a white solid in 30% yield. TLC, LC-MS, 1H NMR and 13C NMR data as for (S,R)-27b, confirming (R,S) configuration of (R)-27b.
Synthesis of (S)-2-((S)-azetidin-2-yl)-2-hydroxyacetic acid (S,S)- 6a.
Obtained from tert-butyl (S)-2-((S)-cyano(hydroxy)methyl)azetidine-1-carboxylate (S,S)-27a (162 mg, 0.76 mmol), according to METHOD B. The residue was crystallized by water/ethanol, providing the desired compound (S,S)-6a in 53% yield. Rt (LC-MS) = 0.516 min. LC-MS (ESI): m/z calcd. for C5H10NO3 [M + H]+= 132.07, found 132.1. Rt (chiral HPLC) = 7.19 min. 1H NMR (600 MHz, D2O) δ 4.85 (dddd, J = 8.9, 8.1, 4.0, 0.8 Hz, 1H), 4.34 (d, J = 4.0 Hz, 1H), 4.09 (ddd, J = 10.6, 9.5, 8.3 Hz, 1H), 3.93 (tdd, J = 10.6, 6.0, 0.8 Hz, 1H), 2.60–2.51 (m, 1H), 2.49–2.42 (m, 1H). 13C NMR (151 MHz, D2O) δ 178.6, 72.8, 64.6, 46.2, 21.7.
Synthesis of (R)-2-((S)-azetidin-2-yl)-2-hydroxyacetic acid (S,R)- 6b.
Obtained from tert-butyl (S)-2-((R)-cyano(hydroxy)methyl)azetidine-1-carboxylate (S,R)-27b (106 mg, 0.50 mmol), according to METHOD B. The residue was crystallized by water/ethanol, providing the desired compound (S,R)-6b in 42% yield. Rt (LC-MS) = 0.581min. LC-MS (ESI): m/z calcd. for C5H10NO3 [M + H]+= 132.07, found 132.0. 1H NMR (600 MHz, D2O) δ 4.82–4.79 (m, 1H), 4.26 (d, J = 4.6 Hz, 1H), 4.13 (qd, J = 9.7, 0.9 Hz, 1H), 3.91 (tdd, J = 10.7, 5.5, 0.9 Hz, 1H), 2.70–2.62 (m, 1H), 2.61–2.53 (m, 1H). 13C NMR (151 MHz, D2O) δ 178.9, 73.7, 65.0, 46.1, 24.1.
Synthesis of (R)-2-((R)-azetidin-2-yl)-2-hydroxyacetic acid (R,R)- 6a.
Obtained from tert-butyl (R)-2-((R)-cyano(hydroxy)methyl)azetidine-1-carboxylate (R,R)-27a (114 mg, 0.54 mmol), according to METHOD B. The residue was crystallized by water/ethanol, providing the desired compound (R,R)-6a in 47% yield. Rt (chiral HPLC) = 8.45 min. LC-MS, 1H NMR and 13C NMR data as for (S,S)-6a, confirming (R,R) configuration of (R,R)-6a.
Synthesis of (S)-2-((R)-azetidin-2-yl)-2-hydroxyacetic acid (R,S)- 6b.
Obtained from tert-butyl (R)-2-((S)-cyano(hydroxy)methyl)azetidine-1-carboxylate (R,S)-27b (117 mg, 0.55 mmol), according to METHOD B. The residue was crystallized by water/ethanol, providing the desired compound (R,S)-6b in 33% yield. LC-MS, 1H NMR and 13C NMR data as for (S,R)-6b, confirming (R,S) configuration of (R,S)-6b.
Synthesis of tert-butyl (S)-2-((S)-cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (S,S)- 28a and of (S)-2-((R)-cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (S,R)- 28b.
Obtained from tert-butyl (S)-2-formylpyrrolidine-1-carboxylate (1.00 g, 5.02 mmol), according to METHOD A. Purification by silica gel flash column chromatography (gradient from DCM to DCM/EtOAc 9:1) allowed separation of the two diastereoisomers (S)-28a and (S)-28b. Diastereoisomer (S)-28a was obtained as a white solid in 31% yield. Rf (DCM/EtOAc 95:5) = 0.56. Rt (LC-MS) = 3.863 min. LC-MS (ESI): m/z calcd. for C7H10N2O3 [M-tBu + H]+= 171.08, found 171.0. 1H NMR (400 MHz, CDCl3) δ 7.07 (d, J = 9.2 Hz, 1H), 4.50 (d, J = 9.2 Hz, 1H), 4.11 (t, J = 7.4 Hz, 1H), 3.64–3.55 (m, 1H), 3.51–3.39 (m, 1H), 2.30–2.16 (m, 1H), 2.10–1.99 (m, 1H), 1.88–1.68 (m, 2H), 1.50 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 158.1, 118.4, 82.2, 67.9, 62.0, 48.4, 29.2, 28.5, 23.8. Mosher analysis determined (S,S) configuration for (S)-28a. Diastereoisomer (S)-28b was obtained as a white solid in 46% yield. Rf (DCM/EtOAc 95:5) = 0.34. Rt (LC-MS) = 3.789 min. LC-MS (ESI): m/z calcd. for C7H10N2O3 [M-tBu + H]+= 171.08, found 171.0. 1H NMR (600 MHz, CDCl3) δ 5.74 (s, 1H), 4.45 (d, J = 8.4 Hz, 1H), 4.19 (t, J = 8.4 Hz, 1H), 3.53–3.42 (m, 1H), 3.42–3.34 (m, 1H), 2.25–2.15 (m, 1H), 2.00–1.84 (m, 3H), 1.47 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 158.0, 118.9, 81.9, 66.7, 61.0, 47.9, 28.5, 28.5, 24.0. 1H NMR analysis determined (S,R) absolute configuration for (S)-27b.
Synthesis of tert-butyl (R)-2-((R)-cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (R,R)- 28a and of (R)-2-((S)-cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (R,S)- 28b.
Obtained from tert-butyl (R)-2-formylpyrrolidine-1-carboxylate (1.00 g, 5.02 mmol), according to METHOD A. Purification by silica gel flash column chromatography (gradient from DCM to DCM/EtOAc 9:1) allowed separation of the two diastereoisomers (R)-28a and (R)-28b. Diastereoisomer (R)-28a was obtained as a white solid in 31% yield. TLC, LC-MS, 1H NMR and 13C NMR data as for (S,S)-28a, confirming (R,R) configuration of (R)-28a. Diastereoisomer (R)-28b was obtained as a white solid in 35% yield. TLC, LC-MS, 1H NMR and 13C NMR data as for (R,R)-28b, confirming (R,R) configuration of (R)-28b.
Synthesis of (S)-2-((S)-pyrrolidin-2-yl)-2-hydroxyacetic acid (S,S)- 7a.
Obtained from tert-butyl (S)-2-((S)-cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (S,S)-28a (91 mg, 0.40 mmol), according to METHOD B. The residue was crystallized from ethanol, providing the desired compound (S,S)-7a in 38% yield. Rt (LC-MS) = 0.551 min. LC-MS (ESI): m/z calcd. for C6H12NO3 [M + H]+= 146.08, found 146.1. Rt (chiral HPLC) = 8.30 min. 1H NMR (600 MHz, D2O) δ 4.33 (d, J = 4.1 Hz, 1H), 3.96 (ddd, J = 9.5, 7.3, 4.1 Hz, 1H), 3.38 (t, J = 7.0 Hz, 2H), 2.15–2.08 (m, 1H), 2.08–1.97 (m, 2H), 1.97–1.86 (m, 1H). 13C NMR (151 MHz, D2O) δ 179.7, 72.2, 64.6, 48.9, 26.7, 26.2.
Synthesis of (S)-2-((S)-pyrrolidin-2-yl)-2-hydroxyacetic acid (S,S)- 7a -TFA.
Prepared by dissolving (S,S)-7a (10 mg, 0.07 mmol, 1 eq) in water (1 mL) and treating it with TFA (6 µL, 0.08 mmol, 1.2 eq) for 10 minutes at room temperature. Evaporation under reduced pressure and freeze drying provided the desired compound (S,S)-7a-TFA as a colorless oil in 77% yield. 1H NMR (600 MHz, MeOD) δ 4.42 (d, J = 4.3 Hz, 1H), 4.02–3.88 (m, 1H), 3.34–3.31 (m, 2H), 2.16–1.92 (m, 4H). 13C NMR (151 MHz, MeOD) δ 174.5, 163.07 (q, J = 34.0 Hz), 117.4 (q, J = 292.2 Hz), 69.7, 62.5, 47.3, 25.5, 24.9.
Synthesis of (R)-2-((S)-pyrrolidin-2-yl)-2-hydroxyacetic acid (S,R)- 7b.
Obtained from tert-butyl (S)-2-((R)-cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (S,R)-28b (88 mg, 0.39 mmol), according to METHOD B. The residue was crystallized from ethanol, providing the desired compound (S,R)-7b in 39% yield. Rt (LC-MS) = 0.544 min. LC-MS (ESI): m/z calcd. for C6H12NO3 [M + H]+= 146.08, found 146.1. 1H NMR (600 MHz, D2O) δ 4.20 (d, J = 5.1 Hz, 1H), 3.84 (ddd, J = 9.2, 7.5, 5.1 Hz, 1H), 3.43–3.31 (m, 2H), 2.27–2.18 (m, 1H), 2.17–2.09 (m, 1H), 2.09–2.01 (m, 1H), 1.97–1.87 (m, 1H). 13C NMR (151 MHz, D2O) δ 179.9, 73.4, 65.1, 48.6, 29.5, 26.3.
Synthesis of (R)-2-((R)-pyrrolidin-2-yl)-2-hydroxyacetic acid (R,R)- 7a.
Obtained from tert-butyl (R)-2-((R)-cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (R,R)-28a (104 mg, 0.46 mmol), according to METHOD B. The residue was crystallized in ethanol, providing the desired compound (R,R)-7a in 55% yield. Rt (chiral HPLC) = 9.08 min. LC-MS, 1H NMR and 13C NMR data as for (S,S)-7a, confirming (R,R) configuration of (R,R)-7a.
Synthesis of (S)-2-((R)-pyrrolidin-2-yl)-2-hydroxyacetic acid (R,S)- 7b.
Obtained from tert-butyl (R)-2-((S)-cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (R,S)-28b (112 mg, 0.50 mmol), according to METHOD B. The residue was crystallized in ethanol, providing the desired compound (R,S)-7b in 41% yield. LC-MS, 1H NMR and 13C NMR data as for (S,R)-7b, confirming (R,S) configuration of (R,S)-7b.
Synthesis of (rac) tert-butyl 2-(cyano(hydroxy)methyl)piperidine-1-carboxylate (rac)- 29a and of (rac)- 29b.
Obtained from tert-butyl (rac)-2-formylpiperidine-1-carboxylate (1.00 g, 4.69 mmol), according to METHOD A. Purification by silica gel flash column chromatography (gradient from DCM to DCM/EtOAc 9:1) allowed separation of the two diastereoisomers (rac)-29a and (rac)-29b. The racemic mixture of diastereoisomer (rac)-29a was obtained as a white solid in 46% yield. Rf (DCM/EtOAc 9:1) = 0.39. Rt (LC-MS) = 3.921 min. LC-MS (ESI): m/z calcd. for C8H13N2O3 [M-tBu + H]+= 185.09, found 185.1.1H NMR (400 MHz, CDCl3) δ 4.65 (d, J = 10.0 Hz, 1H), 4.46 (ddd, J = 9.6, 5.6, 3.0 Hz, 1H), 4.03 (broad s, 1H), 3.90 (m, 1H), 2.90 (m, 1H), 2.03–1.94 (m, 1H), 1.82–1.63 (m, 3H), 1.54–1.48 (m, 2H), 1.47 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 157.7, 119.3, 81.4, 61.7, 54.5, 40.9, 28.4, 24.8, 24.6, 19.5. The racemic mixture of diastereoisomer (rac)-29b was obtained as a white solid in 40% yield. Rf (DCM/EtOAc 9:1) = 0.29. LC-MS (ESI): m/z calcd. for C8H13N2O3 [M-tBu + H]+= 185.09, found 185.1. 1H NMR (400 MHz, CDCl3) δ 5.29 (broad s, 1H), 4.61 (d, J = 5.7 Hz, 1H), 4.37 (q, J = 5.7 Hz, 1H), 4.06–3.87 (m, 1H), 3.22–2.97 (m, 1H), 2.00–1.81 (m, 1H), 1.80–1.56 (m, 5H), 1.49 (s, 9H).13C NMR (151 MHz, CDCl3) δ 155.8, 119.4, 81.3, 61.7, 53.6, 40.8, 28.4, 24.5, 23.8, 18.7.
Synthesis of (rac)-2-(pyrrolidin-2-yl)-2-hydroxyacetic acid (rac)- 8a.
Obtained from (rac)-tert-butyl 2-(cyano(hydroxy)methyl)piperidine-1-carboxylate (rac)-29a (74 mg, 0.31 mmol), according to METHOD B. The residue was crystallized from water/acetone, providing the desired compound (rac)-8a in 47% yield. Rt (LC-MS) = 0.589 min. LC-MS (ESI): m/z calcd. for C7H14NO3 [M + H]+= 160.10, found 160.1. 1H NMR (600 MHz, D2O) δ 4.02 (d, J = 5.4 Hz, 1H), 3.46 (ddt, J = 12.8, 4.2, 2.0 Hz, 1H), 3.33 (ddd, J = 11.7, 5.4, 3.1 Hz, 1H), 3.03 (td, J = 12.8, 3.1 Hz, 1H), 2.03–1.89 (m, 3H), 1.72–1.54 (m, 3H). 13C NMR (151 MHz, D2O) δ 179.6, 75.4, 61.8, 47.8, 28.1, 24.7, 24.2.
Synthesis of (rac)-2-(pyrrolidin-2-yl)-2-hydroxyacetic acid (rac)- 8b.
Obtained from (rac)-tert-butyl 2-(cyano(hydroxy)methyl)piperidine-1-carboxylate (rac)-29b (113 mg, 0.47 mmol), according to METHOD B. The residue was crystallized first from ethanol, and then from methanol/water providing the desired compound (rac)-8b in 24% yield. Rt (LC-MS) = 0.581 min. LC-MS (ESI): m/z calcd. for C7H14NO3 [M + H]+= 160.10, found 160.1. 1H NMR (600 MHz, D2O) δ 4.22 (d, J = 3.5 Hz, 1H), 3.53–3.41 (m, 2H), 3.10 (td, J = 13.0, 3.2 Hz, 1H), 1.99–1.84 (m, 2H), 1.74–1.60 (m, 3H), 1.60–1.49 (m, 1H). 13C NMR (151 MHz, D2O) δ 179.2, 74.3, 61.9, 47.9, 25.1, 24.5, 24.2.
Synthesis of (rac)-tert-butyl 3-(cyano(hydroxy)methyl)azetidine-1-carboxylate (rac)- 32.
Obtained from tert-butyl 3-formylazetidine-1-carboxylate (360 mg, 1.96 mmol), according to METHOD A. Purification by silica gel flash column chromatography (n-heptane/EtOAc 1:1) provided the desired compound (rac)-32 as a white solid in 97% yield. Rf (n-heptane/EtOAc 1:1) = 0.24. 1H NMR (600 MHz, DMSO-d6) δ 6.66 (d, J = 6.4 Hz, 1H), 4.74 (t, J = 6.4 Hz, 1H), 3.93–3.83 (m, 2H), 3.71–3.61 (m, 2H), 2.87 (tdt, J = 8.3, 6.4, 5.4 Hz, 1H), 1.37 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 155.4, 119.8, 78.6, 61.2, 50.7, 49.6, 32.2, 28.0.
Synthesis of 2-(azetidin-3-yl)-2-hydroxyacetic acid (rac)- 9.
Obtained from (rac)-tert-butyl 3-(cyano(hydroxy)methyl)azetidine-1-carboxylate (rac)-32 (200 mg, 0.94 mmol), according to METHOD B. The residue was crystallized from water/methanol, providing the desired compound (rac)-9 in 21% yield. Rt (LC-MS) = 0.526 min. LC-MS (ESI): m/z calcd. for C5H10NO3 [M + H]+= 132.07, found 132.1. 1H NMR (600 MHz, D2O) δ 4.27–4.19 (m, 1H), 4.17–4.06 (m, 4H), 3.35–3.25 (m, 1H). 13C NMR (151 MHz, D2O) δ 181.2, 73.5, 51.1, 49.9, 38.1.
Synthesis of tert-butyl (S)-3-formylpyrrolidine-1-carboxylate (S)- 30.
Obtained from tert-butyl (S)-3-(hydroxymethyl)pyrrolidine-1-carboxylate (1.00 g, 4.97 mmol), according to METHOD C, providing the desired compound (S)-30 in 58% yield. Rf (heptane/EtOAc 1:1) = 0.37. 1H NMR (600 MHz, CDCl3) δ 9.69 (t, J = 1.3 Hz, 1H), 3.77–3.64 (m, 1H), 3.58–3.47 (m, 1H), 3.44–3.29 (m, 2H), 3.02 (p, J = 6.9 Hz, 1H), 2.27–2.15 (m, 1H), 2.15–2.00 (m, 1H), 1.46 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 200.8, 154.5, 79.8, 50.3, 45.2, 45.0, 28.6, 25.8.
Synthesis of tert-butyl (R)-3-formylpyrrolidine-1-carboxylate (R)- 30.
Obtained from tert-butyl (R)-3-(hydroxymethyl)pyrrolidine-1-carboxylate (1.00 g, 4.97 mmol), according to METHOD C, providing the desired compound (R)-30 in 95% yield. TLC, 1H NMR and 13C NMR data as for (S)-30.
Synthesis of tert-butyl (3S)-3-(cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (S)- 33a and (S)- 33b.
Obtained from tert-butyl (S)-3-formylpyrrolidine-1-carboxylate (S)-30 (578 mg, 2.90 mmol), according to METHOD A. Purification by silica gel flash column chromatography (gradient from n-heptane to n-heptane/EtOAc 1:1) allowed separation of the two diastereoisomers (S)-33a and (S)-33b. Diastereoisomer (S)-33a was obtained as a white solid in 16% yield. Rf (n-heptane/EtOAc 1:1) = 0.41. Rt (LC-MS) = 3.616 min. LC-MS (ESI): m/z calcd. for C7H10N2O3 [M-tBu + H]+= 171.08, found 171.0. 1H NMR (600 MHz, CDCl3) δ 4.38 (d, J = 8.2 Hz, 1H), 3.59 (dd, J = 11.5, 7.6 Hz, 1H), 3.55–3.47 (m, 1H), 3.41–3.29 (m, 2H), 2.64 (h, J = 7.6 Hz, 1H), 2.14 (h, J = 7.0 Hz, 1H), 1.91–1.81 (m, 1H), 1.46 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 154.8, 119.3, 80.1, 62.7, 62.4, 47.7, 47.6, 45.3, 45.0, 43.3, 42.7, 28.4, 27.4, 26.7. Diastereoisomer (S)-33b was obtained as a white solid in 27% yield. Rf (n-heptane/EtOAc 1:1) = 0.36. Rt (LC-MS) = 3.596 min. LC-MS (ESI): m/z calcd. for C7H10N2O3 [M-tBu + H]+= 171.08, found 171.0. 1H NMR (400 MHz, CDCl3) δ 4.49 (d, J = 6.7 Hz, 1H), 3.61 (dd, J = 11.3, 7.8 Hz, 1H), 3.53 (ddd, J = 12.1, 8.3, 4.3 Hz, 1H), 3.41–3.27 (m, 2H), 3.08 (bs, 1H), 2.65 (m, 1H), 2.15–2.03 (m, 1H), 2.00–1.87 (m, 1H), 1.46 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 154.9, 119.2, 80.3, 62.6, 62.3, 47.6, 47.2, 45.6, 45.0, 43.3, 42.5, 28.6, 27.0, 26.8.
Synthesis of tert-butyl (3R)-3-(cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (R)- 33a and (R)- 33b.
Obtained from tert-butyl (R)-3-formylpyrrolidine-1-carboxylate (R)-30 (944 mg, 4.74 mmol), according to METHOD A. Purification by silica gel flash column chromatography (gradient from n-heptane to n-heptane/EtOAc 1:1) allowed separation of the two diastereoisomers (R)-33a and (R)-33b. Diastereoisomer (R)-33a was obtained as a white solid in 18% yield. TLC, LC-MS, 1H NMR and 13C NMR data as for (S)-33a. Diastereoisomer (R)-33b was obtained as a white solid in 37% yield. TLC, LC-MS, 1H NMR and 13C NMR data as for (S)-33b.
Synthesis of 2-hydroxy-2-((S)-pyrrolidin-3-yl)acetic acid (S)- 10a.
Obtained from tert-butyl (3S)-3-(cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (S)-33a (81 mg, 0.36 mmol), according to METHOD B. The residue was crystallized from water/ethanol, providing the desired compound (S)-10a in 23% yield. Rt (LC-MS) = 0.583 min. LC-MS (ESI): m/z calcd. for C6H12NO3 [M + H]+= 146.08, found 146.1. 1H NMR (600 MHz, D2O) δ 4.09 (d, J = 4.5 Hz, 1H), 3.46 (ddd, J = 11.3, 8.2, 4.8 Hz, 1H), 3.40–3.28 (m, 2H), 3.22 (dd, J = 11.8, 8.2 Hz, 1H), 2.79 (pd, J = 8.2, 4.5 Hz, 1H), 2.28–2.20 (m, 1H), 1.99 (dq, J = 13.1, 8.2 Hz, 1H). 13C NMR (151 MHz, Deuterium Oxide) δ 182.2, 75.1, 48.8, 48.5, 43.9, 29.7.
Synthesis of 2-hydroxy-2-((S)-pyrrolidin-3-yl)acetic acid (S)- 10b.
Obtained from tert-butyl (3S)-3-(cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (S)-33b (150 mg, 0.66 mmol), according to METHOD B. The residue was decanted twice from water/ethanol, providing the desired compound (S)-10b in 21% yield. Rt (LC-MS) = 0.527 min. LC-MS (ESI): m/z calcd. for C6H12NO3 [M + H]+= 146.08, found 146.1. 1H NMR (600 MHz, D2O) δ 4.09 (d, J = 4.4 Hz, 1H), 3.56–3.40 (m, 2H), 3.34–3.22 (m, 2H), 2.79 (pd, J = 8.1, 4.4 Hz, 1H), 2.12–2.02 (m, 1H), 1.95–1.86 (m, 1H). 13C NMR (151 MHz, D2O) δ 182.2, 74.6, 50.4, 48.6, 43.8, 27.5.
Synthesis of 2-hydroxy-2-((R)-pyrrolidin-3-yl)acetic acid (R)- 10a.
Obtained from tert-butyl (3R)-3-(cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (R)-33a (83 mg, 0.37 mmol), according to METHOD B. The residue was crystallized from water/ethanol, providing the desired compound (R)-10a in 19% yield. LC-MS, 1H NMR and 13C NMR data as for (S)-10a.
Synthesis of 2-hydroxy-2-((R)-pyrrolidin-3-yl)acetic acid (R)- 10b.
Obtained from tert-butyl (3R)-3-(cyano(hydroxy)methyl)pyrrolidine-1-carboxylate (R)-33b (150 mg, 0.37 mmol), according to METHOD B. The residue was decanted twice from water/ethanol, providing the desired compound (R)-10b in 24% yield. LC-MS, 1H NMR and 13C NMR data as for (S)-10b.
Synthesis of tert-butyl (S)-3-formylpiperidine-1-carboxylate (S)- 31.
Obtained from tert-butyl (S)-3-(hydroxymethyl)piperidine-1-carboxylate (1.00 g, 4.64 mmol), according to METHOD C, providing the desired compound (S)-31 in 88% yield. 1H NMR (400 MHz, CDCl3) δ 9.70 (s, 1H), 4.00–3.86 (m, 1H), 3.71–3.59 (m, 1H), 3.32 (dd, J = 13.6, 8.3 Hz, 1H), 3.09 (ddd, J = 13.0, 9.3, 3.4 Hz, 1H), 2.42 (p, J = 4.2 Hz, 1H), 2.01–1.89 (m, 1H), 1.73–1.59 (m, 2H), 1.51–1.48 (m, 1H), 1.46 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 202.7, 154.9, 80.1, 48.2, 44.3, 43.8, 28.6, 24.4, 23.9.
Synthesis of tert-butyl (R)-3-formylpiperidine-1-carboxylate (R)- 31.
Obtained from tert-butyl (R)-3-(hydroxymethyl)piperidine-1-carboxylate (1.00 g, 4.64 mmol), according to METHOD C, providing the desired compound (R)-31 in 71% yield. 1H NMR and 13C NMR data as for (S)-31.
Synthesis of tert-butyl (3S)-3-(cyano(hydroxy)methyl)piperidine-1-carboxylate (S)- 34a and (S)- 34b.
Obtained from tert-butyl (S)-3-formylpiperidine-1-carboxylate (S)-31 (870 mg, 4.08 mmol), according to METHOD A. Purification by silica gel flash column chromatography (gradient from petroleum ether to petroleum ether/diethyl ether 1:1) allowed separation of the two diastereoisomers (S)-34a and (S)-34b. Diastereoisomer (S)-34a was obtained as a white solid in 13% yield. Rf (petroleum ether/diethyl ether 1:1) = 0.35. Rt (LC-MS) = 3.829 min. LC-MS (ESI): m/z calcd. for C8H13N2O3 [M-tBu + H]+= 185.09, found 185.1. 1H NMR (600 MHz, CDCl3) δ 5.11–4.86 (m, 1H), 4.32–4.20 (m, 1H), 4.03–3.93 (m, 1H), 3.86–3.68 (m, 1H), 2.98–2.85 (m, 1H), 2.85–2.72 (m, 1H), 2.02–1.79 (m, 2H), 1.71–1.58 (m, 1H), 1.49–1.35 (m, 11H). 13C NMR (151 MHz, CDCl3) δ 155.3, 119.1, 80.5, 63.1, 45.4, 44.9, 40.2, 28.4, 26.2, 23.9. Diastereoisomer (S)-34b was obtained as a white solid in 15% yield. Rf (petroleum ether/diethyl ether 1:1) = 0.28. Rt (LC-MS) = 3.836 min. LC-MS (ESI): m/z calcd. for C8H13N2O3 [M-tBu + H]+= 185.09, found 185.1. 1H NMR (400 MHz, CDCl3) δ 4.45 (d, J = 5.5 Hz, 1H), 4.14–3.99 (m, 1H), 3.94–3.80 (m, 1H), 3.00–2.82 (m, 2H), 1.99–1.88 (m, 2H), 1.80–1.71 (m, 1H), 1.54–1.48 (m, 2H), 1.47 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 155.1, 118.7, 80.3, 63.4, 45.3, 44.5, 40.2, 28.6, 25.5, 24.1.
Synthesis of tert-butyl (3R)-3-(cyano(hydroxy)methyl)piperidine-1-carboxylate (R)- 34a and (R)- 34b.
Obtained from tert-butyl (R)-3-formylpiperidine-1-carboxylate (R)-31 (705 mg, 3.31 mmol), according to METHOD A. Purification by silica gel flash column chromatography (gradient from petroleum ether to petroleum ether/diethyl ether 1:1) allowed separation of the two diastereoisomers (R)-34a and (R)-34b. Diastereoisomer (R)-34a was obtained as a white solid in 18% yield. TLC, LC-MS, 1H NMR and 13C NMR data as for (S)-34a. Diastereoisomer (R)-34b was obtained as a white solid in 32% yield. TLC, LC-MS, 1H NMR and 13C NMR data as for (S)-34b.
Synthesis of 2-hydroxy-2-((S)-piperidin-3-yl)acetic acid (S)- 11a.
Obtained from tert-butyl (3S)-3-(cyano(hydroxy)methyl)piperidine-1-carboxylate (S)-34a (46 mg, 0.19 mmol), according to METHOD B. The residue was crystallized from water/ethanol, providing the desired compound (S)-11a in 43% yield. Rt (LC-MS) = 0.544 min. LC-MS (ESI): m/z calcd. for C7H14NO3 [M + H]+= 160.10, found 160.1. 1H NMR (400 MHz, D2O) δ 3.99 (d, J = 4.0 Hz, 1H), 3.47–3.36 (m, 1H), 3.33–3.25 (m, 1H), 2.99–2.88 (m, 2H), 2.18 (tt, J = 11.8, 4.0 Hz, 1H), 2.10–1.97 (m, 1H), 1.97–1.86 (m, 1H), 1.86–1.70 (m, 1H), 1.53 (qd, J = 12.6, 3.9 Hz, 1H). 13C NMR (151 MHz, Deuterium Oxide) δ 179.4, 74.5, 45.4, 44.7, 38.0, 25.5, 22.3.
Synthesis of 2-hydroxy-2-((S)-piperidin-3-yl)acetic acid (S)- 11b.
Obtained from tert-butyl (3S)-3-(cyano(hydroxy)methyl)piperidine-1-carboxylate (S)-34b (80 mg, 0.33 mmol), according to METHOD B. The residue was crystallized from water/ethanol, providing the desired compound (S)-11b in 47% yield. Rt (LC-MS) = 0.532 min. LC-MS (ESI): m/z calcd. for C7H14NO3 [M + H]+= 160.10, found 160.1. 1H NMR (600 MHz, D2O) δ 3.99 (d, J = 3.7 Hz, 1H), 3.49–3.44 (m, 1H), 3.44–3.37 (m, 1H), 2.97–2.88 (m, 2H), 2.21 (tq, J = 11.2, 3.7 Hz, 1H), 2.06–1.98 (m, 1H), 1.79–1.66 (m, 2H), 1.50 (qd, J = 12.9, 4.0 Hz, 1H). 13C NMR (151 MHz, D2O) δ 181.7, 76.0, 49.3, 46.9, 40.0, 25.0, 24.5.
Synthesis of 2-hydroxy-2-((R)-piperidin-3-yl)acetic acid (R)- 11a.
Obtained from tert-butyl (3R)-3-(cyano(hydroxy)methyl)piperidine-1-carboxylate (R)-34a (97 mg, 0.40 mmol), according to METHOD B. The residue was crystallized from water/ethanol, providing the desired compound (R)-11a in 30% yield. LC-MS, 1H NMR and 13C NMR data as for (S)-11a.
Synthesis of 2-hydroxy-2-((R)-piperidin-3-yl)acetic acid (R)- 11b.
Obtained from tert-butyl (3R)-3-(cyano(hydroxy)methyl)piperidine-1-carboxylate (R)-34b (120 mg, 0.40 mmol), according to METHOD B. The residue was crystallized from water/ethanol, providing the desired compound (R)-11b in 17% yield. LC-MS, 1H NMR and 13C NMR data as for (S)-11b.
Synthesis of (S)-2-((S)-1-benzylazetidin-2-yl)-2-hydroxyacetic acid (S,S)- 12.
Obtained from (S)-2-((S)-azetidin-2-yl)-2-hydroxyacetic acid (S,S)-6a (30 mg, 0.23 mmol), according to METHOD D. The crude was purified by preparative HPLC followed by ion exchange chromatography, and crystallized from water/ethanol, providing the desired compound in 49% yield. Rt (LC-MS) = 1.488 min. LC-MS (ESI): m/z calcd. for C12H16NO3+ [M + H]+= 222.11, found 222.2. Rt (HPLC) = 6.31 min. 1H NMR (600 MHz, MeOD) δ 7.57–7.50 (m, 2H), 7.50–7.43 (m, 3H), 4.74 (td, J = 8.8, 3.8 Hz, 1H), 4.41 (d, J = 12.9 Hz, 1H), 4.34 (d, J = 12.9 Hz, 1H), 4.05–3.96 (m, 1H), 3.96–3.88 (m, 1H), 3.67–3.54 (m, 1H), 2.47 (dq, J = 11.8, 9.3 Hz, 1H), 2.35–2.22 (m, 1H). 13C NMR (151 MHz, MeOD) δ 174.8, 131.4, 131.3, 131.0, 130.4, 71.4, 70.3, 58.8, 52.3, 18.0.
Synthesis of (S)-2-((S)-1-([1,1'-biphenyl]-3-ylmethyl)azetidin-2-yl)-2-hydroxyacetic acid (S,S)- 13.
Obtained from (S)-2-((S)-azetidin-2-yl)-2-hydroxyacetic acid (S,S)-6a (40 mg, 0.31 mmol), according to METHOD D. The crude was purified by preparative preparative HPLC followed by ion exchange chromatography, and crystallized from water/ethanol providing the desired compound in 32% yield. Rt (LC-MS) = 3.120 min. LC-MS (ESI): m/z calcd. for C18H20NO3+ [M + H]+= 298.14, found 298.2. Rt (HPLC) = 10.37 min. 1H NMR (600 MHz, MeOD) δ 7.79 (t, J = 1.6 Hz, 1H), 7.71 (dt, J = 7.6, 1.6 Hz, 1H), 7.68–7.64 (m, 2H), 7.53 (t, J = 7.6 Hz, 1H), 7.49 (dt, J = 7.6, 1.6 Hz, 1H), 7.45 (t, J = 7.7 Hz, 2H), 7.39–7.34 (m, 1H), 4.74 (td, J = 8.7, 3.8 Hz, 1H), 4.46 (d, J = 12.9 Hz, 1H), 4.39 (d, J = 12.9 Hz, 1H), 4.02–3.95 (m, 1H), 3.93 (td, J = 9.7, 4.6 Hz, 1H), 3.75–3.67 (m, 1H), 2.54–2.42 (m, 1H), 2.34–2.24 (m, 1H). 13C NMR (151 MHz, MeOD) δ 175.0, 143.7, 141.5, 132.2, 130.9, 130.03, 129.99, 129.9, 129.4, 128.9, 128.2, 71.5, 70.5, 58.9, 52.4, 18.0.
Synthesis of (S)-2-((S)-1-([1,1'-biphenyl]-4-ylmethyl)azetidin-2-yl)-2-hydroxyacetic acid (S,S)- 14.
Obtained from (S)-2-((S)-azetidin-2-yl)-2-hydroxyacetic acid (S,S)-6a (40 mg, 0.31 mmol), according to METHOD D. The crude was purified by preparative HPLC followed by ion exchange chromatography, providing the desired compound in 39% yield. Rt (LC-MS) = 3.076 min. LC-MS (ESI): m/z calcd. for C18H20NO3+ [M + H]+= 298.14, found 298.1. Rt (HPLC) = 10.41 min. 1H NMR (600 MHz, DMSO-d6) δ 7.68–7.64 (m, 2H), 7.62 (d, J = 8.2 Hz, 2H), 7.48–7.44 (m, 2H), 7.42 (d, J = 8.2 Hz, 2H), 7.37–7.33 (m, 1H), 4.07 (bs, 2H, exchanges with D2O), 4.03 (d, J = 13.2 Hz, 1H), 3.78 (td, J = 8.2, 3.8 Hz, 1H), 3.68 (d, J = 3.8 Hz, 1H), 3.67 (d, J = 13.2 Hz, 1H), 3.30 (td, J = 8.2, 3.2 Hz, 1H), 3.05 (q, J = 8.2 Hz, 1H), 2.14–2.05 (m, 1H), 1.90 (dtd, J = 10.9, 8.2, 3.2 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 173.0, 139.8, 139.2, 135.8, 129.4, 128.9, 127.4, 126.6, 126.5, 71.1, 68.2, 59.5, 50.5, 18.0.
Synthesis of (S)-2-((S)-1-(4-(benzyloxy)benzyl)azetidin-2-yl)-2-hydroxyacetic acid (S,S)- 15.
Obtained from (S)-2-((S)-azetidin-2-yl)-2-hydroxyacetic acid (S,S)-6a (40 mg, 0.31 mmol), according to METHOD D. The crude was purified by preparative HPLC followed by ion exchange chromatography, providing the desired compound in 57% yield. Rt (LC-MS) = 3.126 min. LC-MS (ESI): m/z calcd. for C19H22NO4+ [M + H]+= 328.15, found 328.2. Rt (HPLC) = 10.73 min. 1H NMR (600 MHz, DMSO-d6) δ 7.44 (d, J = 7.1 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.35–7.29 (m, 1H), 7.28 (d, J = 8.5 Hz, 2H), 6.97 (d, J = 8.5 Hz, 2H), 5.08 (s, 2H), 4.44 (bs, 2H, exchanges with D2O), 3.96 (d, J = 12.8 Hz, 1H), 3.86 (td, J = 8.4, 3.6 Hz, 1H), 3.66 (d, J = 12.8 Hz, 1H), 3.55 (d, J = 3.6 Hz, 1H), 3.36–3.26 (m, 1H), 3.10 (q, J = 8.4 Hz, 1H), 2.13–2.03 (m, 1H), 1.87 (dtd, J = 11.4, 8.4, 3.4 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 172.8, 157.9, 137.1, 130.4, 128.4, 127.8, 127.7, 114.6, 70.7, 69.2, 68.4, 58.6, 50.1, 17.5.
Synthesis of (S)-2-hydroxy-2-((S)-1-(4-hydroxybenzyl)azetidin-2-yl)acetic acid (S,S)- 16.
Obtained from (S)-2-((S)-azetidin-2-yl)-2-hydroxyacetic acid (S,S)-6a (40 mg, 0.31 mmol), according to METHOD D. The crude was purified by preparative HPLC (isocratic 20% B) followed by ion exchange chromatography and crystallization from ethanol/water, providing the desired compound in 31% yield. Rt (LC-MS) = 0.812 min. LC-MS (ESI): m/z calcd. for C12H16NO4+ [M + H]+= 238.11, found 238.2. Rt (HPLC) = 5.25 min. 1H NMR (600 MHz, Deuterium Oxide) δ 7.43 (d, J = 8.5 Hz, 2H), 6.99 (d, J = 8.5 Hz, 2H), 4.82 (td, J = 9.0, 3.7 Hz, 1H), 4.38 (d, J = 13.1 Hz, 1H), 4.33 (d, J = 13.1 Hz, 1H), 4.05 (q, J = 9.8 Hz, 1H), 3.99 (td, J = 9.8, 4.3 Hz, 1H), 3.71–3.60 (m, 1H), 2.54–2.44 (m, 1H), 2.36–2.26 (m, 1H). 13C NMR (151 MHz, Deuterium Oxide) δ 178.0, 159.9, 134.8, 124.0, 119.0, 71.7, 71.6, 59.9, 53.8, 19.3.
Synthesis of (S)-2-((S)-1-(4-acetamidobenzyl)azetidin-2-yl)-2-hydroxyacetic acid (S,S)- 17.
Obtained from (S)-2-((S)-azetidin-2-yl)-2-hydroxyacetic acid (S,S)-6a (40 mg, 0.31 mmol), according to METHOD D. The crude was purified by preparative HPLC followed by ion exchange chromatography, providing the desired compound in 71% yield. Rt (LC-MS) = 1.096 min. LC-MS (ESI): m/z calcd. for C14H19N2O4+ [M + H]+= 279.13, found 279.1. Rt (HPLC) = 5.99 min. 1H NMR (600 MHz, MeOD) δ 7.66 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 8.6 Hz, 2H), 4.72 (td, J = 8.8, 3.8 Hz, 1H), 4.36 (d, J = 13.0 Hz, 1H), 4.29 (d, J = 13.0 Hz, 1H), 4.01–3.94 (m, 1H), 3.91 (td, J = 9.8, 4.6 Hz, 1H), 3.68–3.59 (m, 1H), 2.52–2.42 (m, 1H), 2.32–2.24 (m, 1H), 2.13 (s, 3H). 13C NMR (151 MHz, MeOD) δ 174.9, 171.8, 141.6, 132.0, 126.4, 121.4, 71.2, 70.3, 58.3, 52.1, 23.9, 17.9.
Synthesis of (S)-2-((S)-1-benzylpyrrolidin-2-yl)-2-hydroxyacetic acid (S,S)- 18.
Obtained from (S)-2-hydroxy-2-((S)-pyrrolidin-2-yl)acetic acid (S,S)-7a (40 mg, 0.31 mmol), according to METHOD D. The crude was purified by preparative HPLC (gradient from 0–90% B) followed by ion exchange chromatography, providing the desired compound in 89% yield. Rt (LC-MS) = 0.625 min. LC-MS (ESI): m/z calcd. for C13H18NO3+ [M + H]+= 236.13, found 236.1. Rt (HPLC) = 6.63 min. 1H NMR (600 MHz, CDCl3) δ 7.53–7.47 (m, 2H), 7.41–7.34 (m, 3H), 6.18 (bs, 2H), 4.37 (d, J = 12.7 Hz, 1H), 4.25–4.11 (m, 2H), 3.99–3.78 (m, 1H), 3.43–3.30 (m, 1H), 3.06–2.92 (m, 1H), 2.21–2.13 (m, 1H), 2.13–1.98 (m, 2H), 1.98–1.86 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 174.1, 129.1, 128.3, 127.4, 69.8, 66.6, 57.8, 52.8, 25.2, 22.5.
Synthesis of (S)-2-((S)-1-([1,1'-biphenyl]-3-ylmethyl)pyrrolidin-2-yl)-2-hydroxyacetic acid TFA (S,S)- 19.
Obtained from (S)-2-hydroxy-2-((S)-pyrrolidin-2-yl)acetic acid (S,S)-7a (20 mg, 0.15 mmol), according to METHOD D. The crude was purified by preparative HPLC providing the desired compound as a colorless oil in 42% yield. Rt (LC-MS) = 3.194 min. LC-MS (ESI): m/z calcd. for C19H22NO3+ [M + H]+= 312.16, found 312.2. Rt (HPLC) = 10.60 min. 1H NMR (600 MHz, CDCl3) δ 11.48 (bs, 1H), 7.75–7.73 (m, 1H), 7.71 (dt, J = 7.9, 1.4 Hz, 1H), 7.62–7.58 (m, 2H), 7.54 (t, J = 7.7 Hz, 1H), 7.48–7.43 (m, 3H), 7.41–7.36 (m, 1H), 5.35 (bs, 2H), 4.45 (d, J = 12.9 Hz, 1H), 4.36–4.31 (m, 1H), 4.28 (d, J = 12.9 Hz, 1H), 4.19 (t, J = 7.1 Hz, 1H), 3.76–3.66 (m, 1H), 3.19–3.09 (m, 1H), 2.23–2.02 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 170.7, 143.2, 139.6, 130.3, 129.8, 129.8, 129.6, 129.2, 128.6, 128.3, 127.3, 68.4, 68.2, 59.7, 55.4, 24.6, 23.6.
Synthesis of (S)-2-((S)-1-([1,1'-biphenyl]-4-ylmethyl)pyrrolidin-2-yl)-2-hydroxyacetic acid (S,S)- 20.
Obtained from (S)-2-hydroxy-2-((S)-pyrrolidin-2-yl)acetic acid (S,S)-7a (20 mg, 0.15 mmol), according to METHOD D. The crude was purified by preparative HPLC followed by ion exchange chromatography, and crystallized from water/ethanol, providing the desired compound in 38% yield. Rt (LC-MS) = 3.178 min. LC-MS (ESI): m/z calcd. for C19H22NO3+ [M + H]+= 312.16, found 312.1. Rt (HPLC) = 10.59 min. 1H NMR (600 MHz, DMSO-d6) δ 7.72–7.61 (m, 4H), 7.52–7.43 (m, 4H), 7.38–7.33 (m, 1H), 4.26 (d, J = 13.2 Hz, 1H), 3.93 (d, J = 4.8 Hz, 1H), 3.68 (d, J = 13.2 Hz, 1H), 3.41–3.22 (bs, 2H), 3.16 (dt, J = 8.8, 4.8 Hz, 1H), 2.99–2.93 (m, 1H), 2.53–2.51 (m, 1H), 1.90–1.69 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 174.0, 139.8, 139.3, 136.1, 129.8, 128.9, 127.4, 126.6, 126.6, 69.8, 66.6, 57.4, 52.8, 25.4, 22.5.
Synthesis of tert-butyl (S)-2-(methoxy(methyl)carbamoyl)azetidine-1-carboxylate (S)- 35.
(S)-1-(tert-butoxycarbonyl)azetidine-2-carboxylic acid (5.00 g, 24.9 mmol) and N,O-dimethylhydroxylamine hydrochloride (2.95 g, 30.3 mmol, 1.22 eq ) were dissolved in DMF (50 mL) and the solution was cooled to 0 °C. Upon sequential addition of N-methylmorpholine (3.33 ml, 30.3 mmol, 1.22 eq), HOBt (4.09 g, 30.3 mmol, 1.22 eq), and EDC (4.70 g, 30.3 mmol, 1.22 eq), the reaction mixture was stirred at 0 °C for 2 h and then at room temperature overnight. The mixture was diluted with EtOAc (300 mL), washed with HCl 1M, NaOH 1M and with brine. The organic layer was dried over anhydrous sodium sulfate, filtered and the solvent was evaporated under vacuum, providing the desired product as a colorless oil that solidified upon standing, in 56% yield. Rf (DCM/EtOAc 95:5) = 0.39. Rt (LC-MS) = 3.457 min. LC-MS (ESI): m/z calcd. for C11H21N2O4+ [M + H]+= 245.15, found 245.2. 1H NMR (400 MHz, CDCl3) δ 5.03 (dd, J = 9.0, 5.5 Hz, 1H), 4.05 (dd, J = 9.0, 6.3 Hz, 1H), 3.87 (ddd, J = 9.0, 7.9, 5.5 Hz, 1H), 3.71 (s, 3H), 3.22 (s, 3H), 2.46 (dtd, J = 11.1, 9.0, 6.2 Hz, 1H), 2.12 (ddt, J = 11.1, 9.0, 5.5 Hz, 1H), 1.43 (s, 9H).
Synthesis of tert-butyl (S)-2-acetylazetidine-1-carboxylate (S)- 36.
Under inert atmosphere, intermediate (S)-35 (524 mg, 2.14 mmol) was dissolved in dry THF (12 mL) and cooled to -78 °C. A solution of MeMgCl 3M in THF (1.07 mL, 3.22 mmol, 1.5 eq) was added dropwise and the resulting solution was stirred at the same temperature for 2 h. The reaction mixture was slowly quenched with saturated NH4Claq (25 mL) and extracted with EtOAc (3 × 25 mL). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and the solvent was evaporated under vacuum. The crude was purified by silica gel flash column chromatography (gradient from petroleum ether/AcOEt 9:1 to 8:2), providing the desired product as a white solid in 92% yield. Rf (petroleum ether/AcOEt 9:1) = 0.18. Rt (LC-MS) = 3.655 min. LC-MS (ESI): m/z calcd. for C6H10NO3+ [M + H-tBu]+= 144.07, found 144.1. 1H NMR (400 MHz, CDCl3) δ 4.59 (dd, J = 9.7, 6.1 Hz, 1H), 4.00–3.82 (m, 2H), 2.45 (tdd, J = 11.6, 9.7, 5.9 Hz, 1H), 2.25 (s, 3H), 2.12 (ddt, J = 11.6, 8.8, 6.4 Hz, 1H), 1.43 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 207.7, 156.1, 80.4, 67.0, 47.0, 28.4, 26.1, 19.8.
Synthesis of tert-butyl (2S)-2-(1-cyano-1-hydroxyethyl)azetidine-1-carboxylate (S)- 37a and (S)- 37b.
Obtained from intermediate (S)-36 (380 mg, 1.91 mmol) according to METHOD A. Purification by silica gel flash column chromatography (gradient from DCM to DCM/EtOAc 95:5) allowed separation of the two diastereoisomers (S)-37a and (S)-37b. Diastereoisomer (S)-37a was obtained as a white solid in 83% yield. Rf (DCM/EtOAc 95:5) = 0.82. 1H NMR (600 MHz, CDCl3) δ 6.77 (s, 1H), 4.25 (dd, J = 8.5, 6.7 Hz, 1H), 3.96–3.86 (m, 2H), 2.35–2.27 (m, 1H), 2.20–2.10 (m, 1H), 1.47 (s, 9H), 1.42 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 158.2, 120.1, 82.1, 73.2, 68.8, 46.9, 28.4, 22.4, 19.1. Diastereoisomer (S)-37b was obtained as a white solid in 11% yield. Rf (DCM/EtOAc 95:5) = 0.43. 1H NMR (600 MHz, CDCl3) δ 5.29 (bs, 1H), 4.54 (t, J = 7.8 Hz, 1H), 3.90 (td, J = 8.9, 6.6 Hz, 1H), 3.80 (td, J = 8.9, 5.3 Hz, 1H), 2.43–2.35 (m, 1H), 2.17–2.06 (m, 1H), 1.62 (s, 3H), 1.45 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 157.8, 120.1, 81.9, 70.4, 67.5, 47.0, 28.3, 22.2, 18.5.
Synthesis of 2-((S)-azetidin-2-yl)-2-hydroxypropanoic acid (S)- 21a.
Obtained from intermediate (S)-37a (230 mg, 1.02 mmol) according to METHOD B, providing the desired compound (S)-21a in 53% yield. Rt (LC-MS) = 0.558 min. LC-MS (ESI): m/z calcd. for C6H12NO3+ [M + H]+= 146.08, found 146.1. 1H NMR (600 MHz, Deuterium Oxide) δ 4.75 (t, J = 8.9 Hz, 1H), 4.14–4.06 (m, 1H), 3.86 (td, J = 10.0, 5.2 Hz, 1H), 2.55–2.45 (m, 1H), 2.44–2.36 (m, 1H), 1.37 (s, 3H). 13C NMR (151 MHz, Deuterium Oxide) δ 180.8, 77.0, 68.6, 45.6, 24.4, 22.5.
Synthesis of 2-((S)-azetidin-2-yl)-2-hydroxypropanoic acid (S)- 21b.
Obtained from intermediate (S)-37b (36 mg, 0.16 mmol) according to METHOD B. The residue was crystallized from water/ethanol, providing the desired compound (S)-21b in 57% yield. Rt (LC-MS) = 0.562 min. LC-MS (ESI): m/z calcd. for C6H12NO3+ [M + H]+= 146.08, found 146.1. 1H NMR (600 MHz, Deuterium Oxide) δ 4.84–4.81 (m, 1H), 4.17 (dt, J = 10.3, 9.2 Hz, 1H), 3.90 (td, J = 10.3, 5.3 Hz, 1H), 2.69–2.55 (m, 2H), 1.37 (s, 3H). 13C NMR (151 MHz, Deuterium Oxide) δ 181.4, 77.0, 67.5, 45.6, 23.7, 22.8.
Synthesis of methyl (S)-2-((S)-azetidin-2-yl)-2-hydroxyacetate (S,S)- 38a.
Obtained from (S,S)-6a (110 mg, 0.84 mmol) according to METHOD E as an off-white solid in 85% yield. Rt (LC-MS) = 0.507 min. LC-MS (ESI): m/z calcd. for C6H12NO3+ [M + H]+= 146.08, found 146.1. 1H NMR (600 MHz, MeOD) δ 4.77 (td, J = 8.4, 4.2 Hz, 1H), 4.55 (d, J = 4.2 Hz, 1H), 4.03 (q, J = 9.3 Hz, 1H), 3.92–3.84 (m, 1H), 3.77 (s, 3H), 2.67–2.55 (m, 1H), 2.51–2.41 (m, 1H). 13C NMR (151 MHz, MeOD) δ 171.8, 69.9, 62.0, 53.1, 44.8, 20.5.
Synthesis of methyl (S)-2-hydroxy-2-((S)-pyrrolidin-2-yl)acetate (S,S)- 39a.
Obtained from (S,S)-7a (100 mg, 0.69 mmol) according to METHOD E as an off-white solid in 92% yield. Rt (LC-MS) = 0.535 min. LC-MS (ESI): m/z calcd. for C7H14NO3+ [M + H]+= 160.10, found 160.1.1H NMR (600 MHz, MeOD) δ 4.54 (d, J = 4.2 Hz, 1H), 3.94 (td, J = 7.9, 4.2 Hz, 1H), 3.80 (s, 3H), 3.31–3.27 (m, 2H), 2.14–1.91 (m, 4H). 13C NMR (151 MHz, MeOD) δ 172.7, 69.7, 62.2, 53.1, 47.5, 25.4, 24.9.
Synthesis of methyl (R)-2-hydroxy-2-((S)-pyrrolidin-2-yl)acetate (S,R)- 39b.
Obtained from (S,R)-7b (154 mg, 1.06 mmol) according to METHOD E as an off-white solid in 74% yield. Rt (LC-MS) = 0.650 min. LC-MS (ESI): m/z calcd. for C7H14NO3+ [M + H]+= 160.09, found 160.1. 1H NMR (600 MHz, MeOD) δ 4.37 (d, J = 4.4 Hz, 1H), 3.88 (td, J = 8.2, 4.4 Hz, 1H), 3.82 (s, 3H), 3.30–3.28 (m, 2H), 2.26–2.18 (m, 1H), 2.15–2.08 (m, 1H), 2.07–1.96 (m, 2H). 13C NMR (151 MHz, MeOD) δ 173.0, 70.4, 62.6, 53.1, 47.2, 28.1, 24.9.
Synthesis of methyl (R)-2-hydroxy-2-((R)-pyrrolidin-2-yl)acetate (R,R)- 39a.
Obtained from (R,R)-7a (146 mg, 0.90 mmol) according to METHOD E as an off-white solid in 90% yield. LC-MS, 1H NMR and 13C NMR data as for (S,S)-39a.
Synthesis of methyl (S)-2-hydroxy-2-((R)-pyrrolidin-2-yl)acetate (R,S)- 39b.
Obtained from (R,S)-7b (145 mg, 1.03 mmol) according to METHOD E as an off-white solid in 75% yield. LC-MS, 1H NMR and 13C NMR data as for (S,R)-39b.
Synthesis of methyl (S)-2-hydroxy-2-((S)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)azetidin-2-yl)acetate (S,S)- 40a.
Obtained from (S,S)-38a (50 mg, 0.31 mmol) according to METHOD F as an off-white solid in 37% yield, after purification with preparative HPLC (isocratic 40% B). Rt (LC-MS) = 3.888 min. LC-MS (ESI): m/z calcd. for C31H36NO6+ [M + H]+= 518.25, found 518.3. 1H NMR (600 MHz, MeOD) δ 7.14 (d, J = 15.3 Hz, 1H), 6.97 (d, J = 8.8 Hz, 6H), 6.86–6.82 (m, 6H), 5.25 (dt, J = 15.3, 7.2 Hz, 1H), 4.78 (td, J = 8.6, 3.3 Hz, 2H), 4.37–4.29 (m, 1H), 4.08–3.99 (m, 1H), 3.99–3.94 (m, 1H), 3.94–3.87 (m, 2H), 3.78 (s, 9H), 3.74 (s, 3H), 2.62–2.52 (m, 1H), 2.40–2.30 (m, 1H). 13C NMR (151 MHz, MeOD) δ 171.2, 159.7, 151.0, 138.7, 132.1, 120.0, 114.2, 69.2, 69.0, 60.2, 57.5, 55.7, 53.2, 52.1, 17.1.
Synthesis of methyl (S)-2-hydroxy-2-((S)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)pyrrolidin-2-yl)acetate (S,S)- 41a.
Obtained from (S,S)-39a (80 mg, 0.50 mmol) according to METHOD F as an off-white solid in 19% yield. Rt (LC-MS) = 3.984 min. LC-MS (ESI): m/z calcd. for C32H38NO6+ [M + H]+= 532.27, found 532.2. 1H NMR (600 MHz, MeOD) δ 7.14 (d, J = 15.4 Hz, 1H), 6.99 (d, J = 8.9 Hz, 6H), 6.84 (d, J = 8.9 Hz, 6H), 5.38 (dt, J = 15.0, 7.3 Hz, 1H), 4.57 (d, J = 3.0 Hz, 1H), 4.16–4.07 (m, 1H), 3.97–3.87 (m, 2H), 3.77 (s, 9H), 3.73 (s, 3H), 3.55–3.48 (m, 1H), 3.22–3.14 (m, 1H), 2.11–1.97 (m, 4H). 13C NMR (151 MHz, MeOD) δ 172.2, 159.7, 150.7, 138.7, 132.1, 120.8, 114.2, 68.7, 68.4, 60.2, 57.0, 56.0, 55.7, 53.1, 25.0, 23.4.
Synthesis of methyl (R)-2-hydroxy-2-((S)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)pyrrolidin-2-yl)acetate (S,R)- 41b.
Obtained from (S,R)-39b (51 mg, 0.32 mmol) according to METHOD F as an off-white solid in 25% yield. Rt (LC-MS) = 4.137 min. LC-MS (ESI): m/z calcd. for C32H38NO6+ [M + H]+= 532.27, found 532.3. 1H NMR (600 MHz, MeOD) δ 7.05 (d, J = 15.4 Hz, 1H), 6.97 (d, J = 8.9 Hz, 6H), 6.85 (d, J = 8.9 Hz, 6H), 5.37 (ddd, J = 15.4, 8.3, 6.4 Hz, 1H), 4.31 (d, J = 6.5 Hz, 1H), 4.15–4.06 (m, 1H), 3.89–3.81 (m, 2H), 3.78 (s, 9H), 3.77 (s, 3H), 3.47–3.40 (m, 1H), 3.26–3.15 (m, 1H), 2.34–2.24 (m, 1H), 2.13–2.05 (m, 1H), 2.04–1.94 (m, 2H). 13C NMR (151 MHz, MeOD) δ 172.8, 159.8, 150.6, 138.8, 132.1, 120.9, 114.2, 71.3, 69.2, 60.3, 59.1, 55.7, 55.2, 53.2, 28.7, 23.7.
Synthesis of methyl (R)-2-hydroxy-2-((R)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)pyrrolidin-2-yl)acetate (R,R)- 41a.
Obtained from (R,R)-39a (41 mg, 0.26 mmol) according to METHOD F as an off-white solid in 14% yield. LC-MS, 1H NMR and 13C NMR data as for (S,S)-41a.
Synthesis of methyl (S)-2-hydroxy-2-((R)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)pyrrolidin-2-yl)acetate (R,S)- 41b.
Obtained from (R,S)-39b (49 mg, 0.31 mmol) according to METHOD F as an off-white solid in 22% yield. LC-MS, 1H NMR and 13C NMR data as for (S,R)-41b.
Synthesis of (S)-2-hydroxy-2-((S)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)azetidin-2-yl)acetic acid (S,S)- 22a.
Obtained from (S,S)-40a (35 mg, 0.07 mmol) according to METHOD G as an off-white solid in 74% yield. Rt (LC-MS) = 3.755 min. LC-MS (ESI): m/z calcd. for C30H34NO6+ [M + H]+= 504.24, found 504.2. Rt (HPLC) = 13.30 min. 1H NMR (600 MHz, MeOD) δ 7.14 (d, J = 15.1 Hz, 1H), 6.97 (d, J = 8.8 Hz, 7H), 6.84 (d, J = 8.8 Hz, 7H), 5.26 (dt, J = 15.1, 7.4 Hz, 1H), 4.83–4.76 (m, 1H), 4.27 (d, J = 3.4 Hz, 1H), 4.04 (dd, J = 13.0, 7.4 Hz, 1H), 4.00–3.93 (m, 1H), 3.93–3.87 (m, 2H), 3.78 (s, 9H), 2.62–2.52 (m, 1H), 2.39–2.30 (m, 1H). 13C NMR (151 MHz, MeOD) δ 172.5, 159.8, 150.9, 138.7, 132.1, 120.1, 114.2, 69.8, 69.1, 60.2, 57.5, 55.7, 51.9, 17.2.
Synthesis of (S)-2-hydroxy-2-((S)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)pyrrolidin-2-yl)acetic acid (S,S)- 23a.
Obtained from (S,S)-41a (20 mg, 0.04 mmol) according to METHOD G as a white solid in 70% yield. Rt (LC-MS) = 3.874 min. LC-MS (ESI): m/z calcd. for C31H36NO6+ [M + H]+= 518.25, found 518.2. Rt (HPLC) = 13.45 min. 1H NMR (600 MHz, MeOD) δ 7.13 (d, J = 15.4 Hz, 1H), 6.99 (d, J = 8.9 Hz, 6H), 6.84 (d, J = 8.9 Hz, 6H), 5.39 (dt, J = 15.4, 7.3 Hz, 1H), 4.52 (d, J = 3.1 Hz, 1H), 4.13 (dd, J = 13.3, 7.3 Hz, 1H), 3.97–3.91 (m, 2H), 3.78 (s, 9H), 3.55–3.47 (m, 1H), 3.22–3.15 (m, 1H), 2.14–1.96 (m, 4H). 13C NMR (151 MHz, MeOD) δ 173.3, 159.8, 150.6, 138.7, 132.1, 120.8, 114.2, 69.0, 68.1, 60.2, 56.9, 56.0, 55.7, 25.0, 23.3.
Synthesis of (R)-2-hydroxy-2-((S)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)pyrrolidin-2-yl)acetic acid (S,R)- 23b.
Obtained from (S,R)-41b (38 mg, 0.07 mmol) according to METHOD G as a white solid in 45% yield. Rt (LC-MS) = 4.473 min. LC-MS (ESI): m/z calcd. for C31H36NO6+ [M + H]+= 518.25, found 518.3. Rt (HPLC) = 13.68 min. 1H NMR (600 MHz, MeOD) δ 7.05 (d, J = 15.4 Hz, 1H), 6.97 (d, J = 8.9 Hz, 6H), 6.84 (d, J = 8.9 Hz, 6H), 5.38 (ddd, J = 15.4, 8.3, 6.3 Hz, 1H), 4.27 (d, J = 6.2 Hz, 1H), 4.13 (dd, J = 13.2, 6.3 Hz, 1H), 3.88–3.80 (m, 2H), 3.78 (s, 9H), 3.49–3.41 (m, 1H), 3.23–3.16 (m, 1H), 2.35–2.26 (m, 1H), 2.12–1.97 (m, 3H). 13C NMR (151 MHz, MeOD) δ 174.4, 160.0, 150.8, 139.0, 132.4, 121.2, 114.5, 71.2, 69.8, 60.6, 59.1, 56.0, 55.4, 29.1, 23.9.
Synthesis of (R)-2-hydroxy-2-((R)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)pyrrolidin-2-yl)acetic acid (R,R)- 23a.
Obtained from (R,R)-41a (19 mg, 0.04 mmol) according to METHOD G as a white solid in 32% yield. HPLC, LC-MS, 1H NMR and 13C NMR data as for (S,S)-23a.
Synthesis of (S)-2-hydroxy-2-((R)-1-((E)-4,4,4-tris(4-methoxyphenyl)but-2-en-1-yl)pyrrolidin-2-yl)acetic acid (R,S)- 23b.
Obtained from (R,S)-41b (35 mg, 0.07 mmol) according to METHOD G as a white solid in 66% yield. HPLC, LC-MS, 1H NMR and 13C NMR data as for (S,R)-23b.
Synthesis of (E)-4,4',4''-(4-bromobut-2-ene-1,1,1-triyl)tris(methoxybenzene) 42.
4,4',4''-(chloromethanetriyl)tris(methoxybenzene) (5.00 g, 13.36 mmol) was dissolved in acetonitrile (125 ml) and stirred vigorously. A solution of NaOH (1M, 20 ml, 1.5 eq) was added dropwise and the reaction mixture was stirred at 40 °C overnight. Upon dilution with water (30 ml), the acetonitrile was evaporated under vacuum, and the aqueous solution was extracted with diethyl ether (5 × 15 mL). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and the solvent was removed under vacuum, providing tris(4-methoxyphenyl)methanol as a viscous orange oil in 98% yield, that was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.17 (d, J = 8.8 Hz, 6H), 6.83 (d, J = 8.8 Hz, 6H), 3.80 (s, 9H), 2.65 (bs, 1H). 13C NMR (101 MHz, CDCl3) δ 158.8, 139.9, 129.2, 113.3, 81.3, 55.4. The crude (4.65 g, 12.61 mmol) and allyltributyltin (8.13 mL, 8.77 g, 26.48 mmol, 2.1 eq) were dissolved in DCM (40 mL), and BF3-OEt2 (3,27 mL, 3.76 g, 26.48 mmol, 2.1 eq) was added dropwise. The reaction mixture was stirred at room temperature for 6 hours. Upon quenching by addition of a saturated solution of NaHCO3, the aqueous layer was extracted three times with DCM. The combined organic layers were dried over anhydrous sodium sulfate, filtered and the solvent was evaporated under reduced pressure, providing a crude that was purified by flash column chromatography (heptane/EtOAc 7:3 + 5% TEA). 4,4',4''-(but-3-ene-1,1,1-triyl)tris(methoxybenzene) was obtained as a white solid in 83% yield. 1H NMR (600 MHz, CDCl3) δ 7.09 (d, J = 8.9 Hz, 6H), 6.79 (d, J = 8.9 Hz, 6H), 5.67 (ddt, J = 17.2, 10.3, 6.6 Hz, 1H), 5.02 (dq, J = 17.2, 1.5 Hz, 1H), 4.94 (dq, J = 10.3, 1.5 Hz, 1H), 3.78 (s, 9H), 3.34 (dt, J = 6.6, 1.5 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 157.6, 140.2, 136.5, 130.4, 117.2, 113.1, 55.3, 54.4, 46.2. The intermediate 4,4',4''-(but-3-ene-1,1,1-triyl)tris(methoxybenzene) (3.00 g, 8.01 mmol, 1 eq) was dissolved in CCl4 (30 mL), and NBS (1.43 g, 8.01 mmol, 1 eq) and AIBN (79 mg, 0.48 mmol, 0.06 eq) were added. The reaction mixture was refluxed overnight and controlled by NMR. The suspension was filtered, and the filtrate was evaporated under vacuum. The residue was purified by flash column chromatography (gradient from heptane to heptane/EtOAc 9/1) providing the desired product 42 as a viscous pale yellow oil in 66% yield. 1H NMR (400 MHz, CDCl3) δ 6.96 (d, J = 8.9 Hz, 6H), 6.81 (d, J = 8.9 Hz, 7H), 6.72 (dd, J = 15.3, 1.0 Hz, 1H), 5.52 (dt, J = 15.3, 7.7 Hz, 1H), 4.09 (dd, J = 7.7, 1.0 Hz, 2H), 3.80 (s, 9H).
{kind=link}
{kind=link}
{kind=link}